
K. Önnestam1, B. Nilsson1, M. Rother1, E. Rein-Hedin2,3, J. Bylund2, P. Anderer4, M. Kemethofer4, M.M. Halldin1, J. Sandin1,5, M. Segerdahl1,5
1. AlzeCure Pharma AB, Hälsovägen 7, SE-14157 Huddinge, Sweden; 2. CTC Clinical Trial Consultants AB, Dag Hammarskjölds väg 10B, SE-75237 Uppsala, Sweden; 3. Department of Surgical Sciences, Plastic Surgery, Uppsala University, SE-75185 Uppsala, Sweden; 4. The Siesta Group Schlafanalyse GmbH, Schlosshofer Strasse 11-3rd Floor, A-1210 Vienna, Austria; 5. Karolinska Institute, Department of Neurobiology, Care Sciences and Society, Alfred Nobels allé 23, SE-141 52 Huddinge, Sweden
Corresponding Author: Märta Segerdahl, AlzeCure Pharma AB, Hälsovägen 7, SE-141 57 Huddinge, Sweden, Phone: +46 736 808 898, E-mail: marta.segerdahl@alzecurepharma.com
J Prev Alz Dis 2023;4(10):778-789
Published online July 12, 2023, http://dx.doi.org/10.14283/jpad.2023.89
Abstract
BACKGROUND: ACD856 is a positive allosteric modulator of tropomyosin receptor kinase (Trk) receptors which has shown to have pro-cognitive and anti-depressant-like effects in various animal models. It is currently in clinical development for the treatment of Alzheimer’s disease and other disorders where cognition is impaired and is also considered for indications such as depression or other neuropsychiatric diseases. ACD856 has a novel mechanism of action modulating the activity of the Trk-receptors, resulting in increased stimulation of the neurotrophin signaling pathways. Previous studies applying single intravenous and oral doses of ACD856 indicate that ACD856 is safe and well-tolerated by healthy volunteer subjects, and that it has suitable safety and pharmacokinetic properties for further clinical development.
OBJECTIVES: To investigate the safety and tolerability of 7 days of treatment with multiple ascending oral doses of ACD856 in healthy subjects, and to characterize its pharmacokinetic (PK) properties. In addition, pharmacodynamic effects of ACD856 using quantitative electroencephalography (qEEG) as an indicator for central target engagement were assessed.
DESIGN: This was a prospective, phase I, double-blind, parallel-group, placebo-controlled, randomized study of the safety, tolerability, PK and pharmacodynamics of multiple ascending oral doses of ACD856 in healthy subjects. ACD856 or placebo were administered in 3 ascending dose cohorts of 8 subjects. Within each cohort, subjects were randomized to receive either ACD856 (n=6) or placebo (n=2).
SETTING: The study was conducted at a First-in-Human unit in Sweden.
PARTICIPANTS: Twenty-four healthy male and female subjects.
INTERVENTION: The study medication was administered as an oral solution, with ACD856 or the same contents without the active ingredient (placebo). The dose levels ranged from 10 mg to 90 mg. ACD856 was administered once daily for 7 days, targeting steady state.
MEASUREMENTS: Safety and tolerability assessments included adverse events, laboratory, vital signs, 12-lead electrocardiogram (ECG), physical examination, assessment of stool frequency and questionnaires to assess symptoms of anxiety, depression, as well as suicidal ideation and behavior. In addition, cardiodynamic ECGs were extracted to evaluate cardiac safety. PK parameters were calculated based on measured concentrations of ACD856 in plasma, urine, and cerebrospinal fluid (CSF) samples. Metabolite profiling, characterization and analysis was performed based on and urine samples. qEEG was recorded for patients in the two highest dose cohorts (30 and 90 mg/day) as a pharmacodynamic assessment to explore central target engagement.
RESULTS: Treatment with ACD856 was well tolerated with no serious adverse events. No treatment emergent or dose related trends were observed for any of the safety assessments. ACD856 was rapidly absorbed and reached maximum plasma exposure at 30 to 45 minutes after administration. Steady state was reached before Day 6, with an elimination half-life at steady state of approximately 20 hours. At steady state, ACD856 exhibited accumulation ratios for Cmax and AUC of approximately 1.6 and 1.9 respectively. The exposure, Cmax and AUC0-24, increased proportionally with the dose. There was no unchanged ACD856 detected in urine. The metabolic pattern in urine and plasma was similar, and in alignment with the metabolites observed in preclinical toxicology studies. The level of ACD856 measured in CSF at steady state increased with dose, indicating Central Nervous System (CNS) exposure at relevant levels for pharmacodynamic effects. ACD856 demonstrated significant dose-dependent treatment-associated changes on qEEG parameters. Specifically, increase of the relative theta power and decrease of the fast alpha and beta power was observed, leading to an acceleration of the delta+theta centroid and an increase in the theta/beta ratio.
CONCLUSIONS: ACD856 was well tolerated at the tested dose levels (10-90 mg/daily for 7 days) in healthy subjects. The compound has a robust pharmacokinetic profile, with rapid absorption and dose-dependent exposure. ACD856 was shown to pass the blood-brain-barrier, reach relevant exposure in the CNS and to induce dose-dependent treatment-related changes on qEEG parameters, indicating central target engagement.
Key words: Alzheimer’s Disease, ACD856, neurotrophin, multiple ascending doses, Trk.
Introduction
The number of individuals living with Alzheimer’s disease (AD) worldwide is constantly growing as an increasing part of the human population is reaching an older age. In the United States (US), it is estimated that in 2022, 6.5 million people had symptoms of dementia due to AD, and at least 5 million are living with mild cognitive impairment due to AD (1). Developing safe and effective treatments for AD has been challenging. More than one hundred pharmaceuticals are in clinical development for this disease, where the majority (83%) of treatments are attempting to alter the progression of the disease (disease modifying), and 10% representing treatments aiming to provide symptomatic cognitive enhancement (2).
To date, treatment options available to patients with mild to moderate AD have been restricted to symptomatic treatment with acetylcholinesterase inhibitors and memantine, with common side effects and limited efficacy (3). Recently, substantial progress has been made in the development of disease modification agents targeting the amyloid cascade (4, 5, 6) with the US Food and Drug Administration recently granting approvals for the antibodies aducanumab and lecanemab, as well as the expected filing of donanemab during 2023. However, it remains to be seen how commonly the antibodies will be used in clinical practice. The medical need for novel effective and safe symptomatic treatments as well as for disease modification, will likely continue to grow.
The neurotrophins nerve growth factor (NGF) and brain derived neurotrophic factor (BDNF) mediate their effects by binding to the tropomyosin receptor kinase receptors TrkA and TrkB, respectively. Trk-receptor mediated signaling pathways play an important role in cell function, cell communication, and cell survival in brain areas vital for cognitive function, such as the hippocampus and basal forebrain (7, 8). There is evidence suggesting that loss of neurotrophin signaling, or decreased levels of neurotrophins in the brain and the cerebrospinal fluid, is associated with disease states characterized by cognitive decline such as AD (9, 10, 11, 12, 13). The rationale for developing treatments aiming to modulate the activity of the Trk receptor signaling relies on evidence suggesting that increased BDNF signaling could lead to improved cognition in AD (14, 15, 16). Interestingly, neurotrophins and their signaling pathways also play a role in depression. A review of clinical evidence concludes that BDNF plays a part in the neurogenesis and adaptation of neuronal networks involved in mood regulation, and that neurotrophins, by regulating transmitter release, induce effects as seen with anti-depressant treatments (17). It has even been suggested that various classes of antidepressants exert their effect directly or indirectly through TrkB receptor-mediated signaling (18).
The gene encoding for BDNF is located on chromosome 11q13. In about 30% of the European population (19), this location contains a functional nucleotide polymorphism (rs6265) which causes a valine (Val) to methionine (Met) amino acid substitution at codon 66 of pro-BDNF (Val66Met). The Met variant is associated with impaired dendritic trafficking of BDNF and decreased secretion of BDNF (18 to 30% decrease) (19). Interestingly, genetic research shows that carriers of the Val66Met-BDNF polymorphism appear to be susceptible to a more rapid deterioration in memory and hippocampal function in disease states such as AD compared to the general population (20, 21). The Val66Met-BDNF polymorphism has also been associated with cognitive impairment in individuals suffering from Parkinson’s disease (22). Moreover, BDNF dysfunction seems to be involved in the cognitive impairment related to sleep disorders (23). The importance of the Val66Met-BDNF polymorphism in relation to sleep intensity and its potential restorative function has also been studied using electroencephalogram (EEG) analysis (24). EEG data in healthy volunteers suggests that the Val66Met-BDNF polymorphism is associated with a reduced network activity pattern compared to non-carriers (25, 26). Investigations of the effect of the Val66Met polymorphism suggest that electrophysiological methods, such as resting EEG, can be used as BDNF sensitive functional markers in early clinical development to examine target engagement (25).
ACD856 is a small molecule, presently in clinical development for the treatment of AD and other disorders where cognition is impaired. Through its novel mechanism of action, ACD856 mediates positive allosteric modulation of Trk-receptors, resulting in enhanced activity of neurotrophin signaling pathways (27). It is hypothesized that positive allosteric modulators, such as ACD856, have treatment advantages compared to a receptor antagonist, given its requirement of the endogenous ligand being present thus having a more spatially and temporally restricted activation pattern. This would potentially provide for a more physiologically relevant pattern of signal enhancement as well as an improved safety profile. Pro-cognitive effects of ACD856 have been observed both in mouse models of induced memory impairment, and in aged animals having a natural decline in memory (28). Repeated dosing of low doses of ACD856 reversed a scopolamine-induced memory impairment in mice as efficiently as a single higher dose.
ACD856 is also being considered for clinical development in indications such as depression or other psychiatric diseases since it has shown anti-depressant like effects in various rodent models, in addition to its pro-cognitive effects (27, 28). Following repeated dosing of ACD856 in animals, a sustained anti-depressant like effect up to three days after the last day of treatment was observed, suggestive of long-term plastic network changes that maintain efficacy over time (29). The study, conducted in the forced swim test in mice, included the positive reference compound ketamine. Ketamine has been reported to induce long-term effects after a single administration and recent data indicate that it mediates its anti-depressant effect through BDNF/TrkB-receptor activation (18). In fact, a similar effect size was observed with ketamine and ACD856 in this preclinical study. Taken together, the data available to date supports clinical development of ACD856 in depression and cognitive disorders based on the hypothesis of stimulated neurotrophin signaling to lead to increased network connectivity in the brain.
ACD856 has previously been evaluated in healthy subjects following single ascending oral doses of 1 to 150 mg (SAD study, NCT05077631, 30). ACD856 was well tolerated with no reports of severe or serious adverse events, and there were no treatment emergent or dose related trends for adverse events (AEs), laboratory safety assessments, vital signs, 12-lead electrocardiogram (ECG), physical examination, or stool frequency. A review of publications from clinical studies using treatment approaches with similar modes of action (enhancing the NGF or BDNF signaling (31, 32, 33, 34, 35, 36, 37) identified changes of gut motility (increased frequency of stools and loose stools), diffuse myalgia, reduced appetite and weight loss, and eosinophilia as potential target related adverse effects. None of these had been described as severe or serious adverse events. Safety pharmacology and toxicology studies of ACD856 have not demonstrated any safety findings, e.g. changes to body weight/body weight gain, food consumption, ophthalmology, coagulation parameters, urinalysis parameters ECG, hematology-, clinical chemistry-, urinalysis- or coagulation parameters at highest dose levels tested up to one month in rat and minipig.
This paper describes the next step in clinical development where the safety and tolerability of multiple ascending oral doses of ACD856 was evaluated in healthy subjects (NCT05077501). Evaluation of its pharmacokinetic (PK) properties was defined as a secondary objective. The study also explored treatment-induced pharmacodynamic effects of ACD856 as measured with quantitative EEG.
Methods
Study Design
This multiple ascending dose (MAD) study was a prospective, phase I, double-blind, parallel-group, placebo-controlled, randomized study to evaluate the safety, tolerability, PK and pharmacodynamics of three multiple ascending doses (10 mg, 30 mg, and 90 mg) of ACD856 administered orally to healthy subjects. ACD856 or placebo was administered in ascending dose cohorts comprising of 8 subjects randomized to receive ACD856 (n=6) or placebo (n=2). Subjects received ACD856 or placebo once daily for 7 days, aiming to reach steady state during multiple dosing. The dose levels were set in accordance with the EMA guidance on First-in-Human (FIH) trials (38) and targeting a range which included the assumed pharmacologically active dose which was estimated based on the observed exposure in the mouse cognition passive avoidance model which had a demonstrated pharmacological effect (28), and the No-Adverse-Effect-Level (NOAEL) in the safety studies.
Dosing was done in a sentinel fashion with the start of dosing staggered, in accordance with available guidance for FIH clinical trials (38). The first two subjects in each MAD cohort (one ACD856 and one placebo), were dosed during the same day and were carefully monitored by clinical staff during and after dosing including regular checks of vital signs and ECG parameters. At least 48 hours after administration of the first dose to the sentinel subjects were allowed for the observation of any reactions before initiating dosing of the remaining subjects. These subjects were enrolled in groups of two or three and dosing was initiated with at least 24 hours between each group. See Figure 1a for a schematic overview of the cohorts and dose groups within each cohort. There was immediate access to equipment, qualified staff, and an intensive care unit in case of an emergency After the completion of each cohort, the safety, tolerability, and PK of ACD856 were assessed by a Safety Review Committee which decided on escalation of the dose to the next cohort.
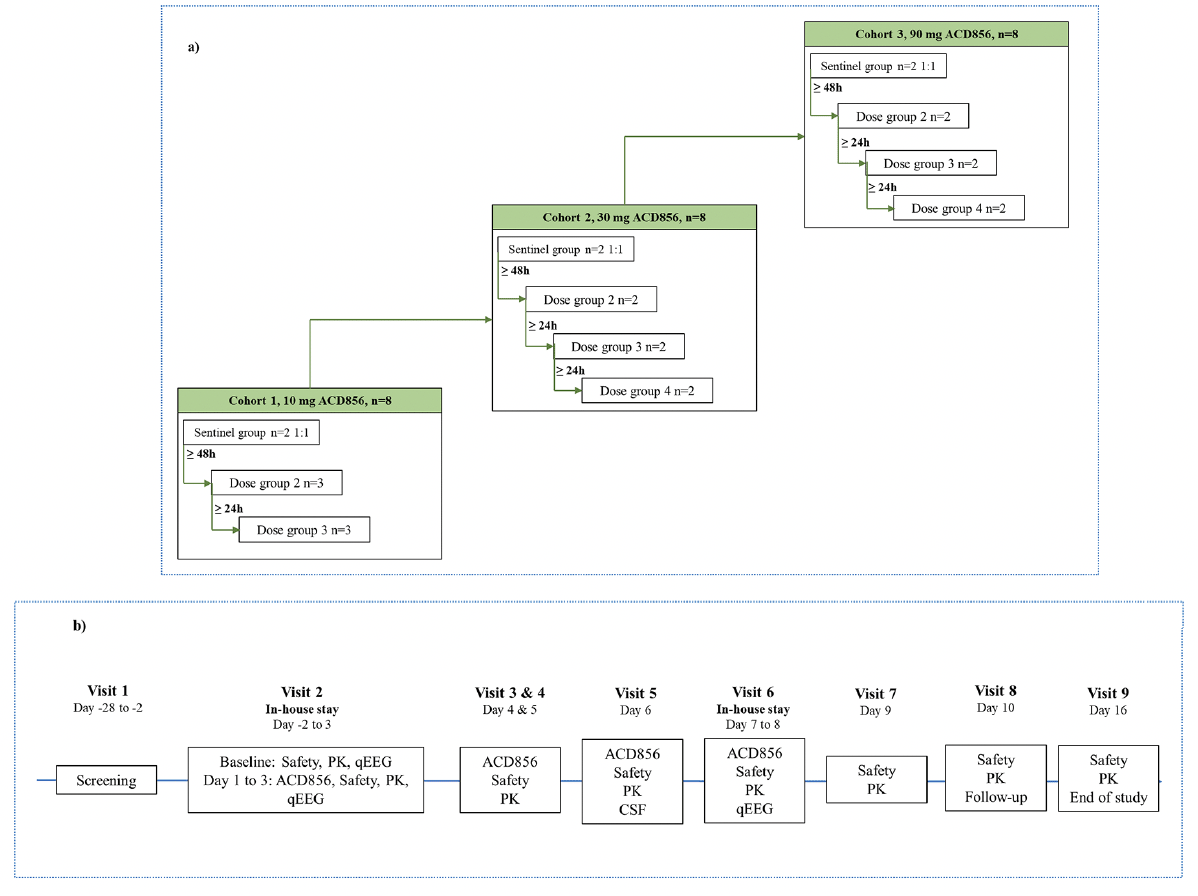
Figure 1. Schematic overview of the multiple ascending dose study
1a: Illustrates the dose escalation between the three cohorts of the multiple ascending dose study with ACD856 including how the initiation of dosing was performed within each cohort. n=number of participating subjects; 1b: Provides a schematic overview of the assessments performed at each study visit of the multiple ascending dose study with ACD856 as well as which visits were performed as in-house stay at the phase I unit. PK=pharmacokinetics, qEEG=quantitative electroencephalography
The study medication was administered orally as a clear solution with ACD856 (2 mg/mL) or the same contents without the active ingredient (placebo). An unblinded pharmacist prepared the study medication on site in a separate room, marked them with subject number and then provided the medication to the study staff. All other site staff, study subjects, monitor and sponsor personnel were successfully blinded to study treatment allocation until after database lock.
The study was performed at the FIH unit of CTC Clinical Trial Consultants AB, Uppsala, Sweden. Subjects were recruited via local advertisements. Verbal and written informed consent were obtained prior to any study related activities.
Study Participants
Study subjects were eligible for inclusion if they were healthy males, or healthy females of non-childbearing potential, aged ≥18 and ≤65 years, and with a body mass index between 18.0 and 30.0 kg/m2 at screening.
Study Assessments
Safety and tolerability assessments included collection of adverse events (AEs), laboratory (clinical chemistry, hematology, coagulation, urinalysis, refer to supplementary table S1), vital signs (systolic and diastolic blood pressure, pulse rate and body temperature), 12-lead ECG and physical examination. Additionally, stool frequency was assessed as changes in gut motility (increased frequency of stools and loose stools) is a known potentially target related AE reported from treatment approaches with similar modes of action, as described in the introduction. AEs were collected from the start of study treatment until the end-of-study visit. Severity grading of AEs were done according to Common terminology criteria for adverse events (CTCAE) v5.0 (39). Causality of the AEs were assessed, on blinded data, evaluating whether there was a reasonable possibility that the event may have been caused by the study treatment. For further assessment of cardiac safety, ECGs were extracted from Holter recordings at baseline and following ACD856 dosing, and were analyzed by Nabios GmbH, in Munich, Germany.
The study also included questionnaires to assess symptoms of anxiety (Generalized Anxiety Disorder-7 scale (40)) and depression (Patient Health Questionnaire-9 (41)), as well as suicidal ideation and behavior (Colombia Severity Suicidal Rating Scale (42)).
Blood and urine samples were collected at pre-defined timepoints in relation to study treatment throughout the study for the determination of ACD856 concentrations and for calculation of PK parameters. Blood samples were taken on Day 1 prior to dosing (0 h) and at 0.17, 0.25, 0.33, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 8 and 12 h after administration. On Days 2, 3, 4, 5 and 6, only pre-dose PK blood samples were taken within 15 min before IMP administration. For the last dose, administered on Day 7, PK blood sampling occurred pre-dose and at 0.17, 0.25, 0.33, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 48 and 72 h post-dose and finally at the end of study visit on Day 16. The blood samples were centrifuged at 1500G for 10 minutes to separate the plasma. The plasma samples were frozen within 1 hour after collection and maintained frozen (at ≤ ‑70°C) until bioanalysis. Urine samples were collected on Day 1 and Day 7 during the following intervals: pre-dose, 0-6, 6-12 and 12-24 hours, and stored frozen (at ≤ ‑70°C) until analysis.
A lumbar puncture was performed on Day 6, 1.5 hours post-dose, to allow for collection of cerebrospinal fluid (CSF) and stored frozen (at ≤ ‑70°C) until analysis.
The concentrations of ACD856 in plasma, urine and CSF were analyzed by Lablytica Life Science AB, Sweden. The bioanalysis method of plasma and urine were validated in accordance with the current EMA guideline on bioanalytical method validation (43) prior to study initiation. The robustness of the bioanalysis method in CSF was qualified through assessing the parallelism of human CSF and synthetic CSF in QC samples of different levels. The bioanalytical method utilized extraction of ACD856 followed by liquid chromatography–mass spectrometry/mass spectrometry (LC‑MS/MS) analysis. The lower limit of quantification (LLOQ) for plasma was set to 10 ng/mL, 3.0 ng/mL for CSF and 100 ng/mL for urine.
The MIST (Metabolite in Safety Testing) analysis and metabolite profiling in plasma and urine was conducted by MetaSafe Sweden AB, Sweden. The MIST analysis was performed in accordance with guidelines on Metabolite in Safety Testing (MIST) (44, 45). Plasma AUC0-24h pools were prepared through matrix matching where the AUC0-24h pool of each cohort on Day 7 was matched with blank plasma from the two toxicology species (rat and minipig, mixed gender). The matrix matched AUC0-24h pools were prepared for analysis by protein precipitation using acetonitrile containing internal standard. The samples were centrifuged, and supernatants transferred to LC vials for LC-(HR)MS analysis. The human metabolite profile in plasma and urine was established based on samples for the 0-24 h time interval from MAD cohort 3 (90 mg/day, Day 7). The samples were prepared by centrifugation and supernatants analyzed by LC-high resolution MS and metabolites searched for by extracted ion chromatogram using m/z of molecular ions [M+H]+ or [M-H]. In addition, software-aided data mining was used (Compound DiscovererTM 3.2, Thermo Scientific, USA) including structure-intelligent dealkylation tool and mass defect filter with manual confirmation. Structures of metabolites were proposed based on accurate mass and fragment ion data when available.
qEEG was recorded for the two highest dose cohorts (30 and 90 mg/day) to enable assessment of pharmacodynamic central nervous system (CNS) target engagement. EEG recordings were performed at baseline, 1.5, 6 and 24 hours following the first dose of study treatment, and at 1.5 hours following the last dose on Day 7. qEEG was recorded from 21 electrodes positioned in accordance with the international 10/20 system and evaluated according to Jobert et al. 2012 (46). Three different recording conditions were applied for each timepoint; vigilance-controlled EEG recording with eyes closed, resting EEG recording with eyes closed, and resting EEG recording with eyes open. qEEG recordings were analyzed by the Siesta Group in Austria. Absolute and relative power as well as frequency measures (centroids and their deviations) were determined for the frequency bands as recommended by the International Pharmaco-EEG Society (46). Derived variables included the alpha slow-wave index (ASI) and the theta/beta ratio (TBR).
A high-level overview of the study design and assessments is shown in Figure 1b.
Pharmacokinetic and Statistical Analysis
No formal sample size calculations were performed. The sample size was considered sufficient to provide adequate information for the study objectives as per current guidance. Safety and PK data were summarized by descriptive statistics. Dose proportionality was analyzed using linear regression modelling. All descriptive summaries and statistical analyses were performed using SAS Version 9.4.
The cardiac safety ECG data analysis was performed based on cardiac intervals and heart rates calculated automatically by the 12SL algorithm (GE Healthcare, Chicago, Illinois, USA). The extracted ECGs were imported into the ECG study database and processed electronically. The assessed cardiac intervals were RR, QT, QRS, PR(PQ), and QTcF.
The PK parameters were calculated by non-compartmental analysis using the software Phoenix WinNonlin® version 8.3 (Certara, U.S.A.). Plasma concentrations below the LLOQ were set to 0 before time to peak concentration (tmax) and to missing thereafter. The area under the curve (AUC) was calculated based in actual timepoints and according to the linear up- log down method. Pharmacokinetic properties of ACD856 on Day 1 included maximum plasma concentration (Cmax), elimination half-life (t1/2(z)), area under the concentration-time curve during a dosing interval (tau) (AUC0–24/tau), apparent plasma clearance (CL/F), and the apparent volume of distribution (Vz/F). Dose proportionality based on AUC0–24 and Cmax was assessed. On Day 7 PK parameters were established for Cmax, tmax, t1/2(z), apparent plasma clearance at steady state (CLss/F), Vz/F, and accumulation ratio (AR) for Cmax and AUCtau between Days 7 and 1. Dose proportionality based on AUCtau and Cmax was assessed at steady state. CSF ACD856 concentrations at steady state (Day 6) were determined, as well as trough plasma concentrations of ACD856 on Days 2-8 (Ctrough).
qEEG analysis was performed in a stepwise approach. Spectral analysis was completed prior to database lock, followed by a statistical analysis of the variables derived from spectral density to evaluate treatment induced changes. The latter part was completed after study unblinding. Paired samples t-tests were applied for post-pre changes within each treatment group. Independent samples t-tests were used to compare post-pre changes between the treatment groups.
Results
Study Population
Twenty-four healthy subjects were randomized and dosed. All enrolled subjects completed 7 days of treatment with study medication. All except one subject attended all study visits; one follow-up visit was missed due to an AE (nasopharyngitis). The study population consisted of 21 males and three females with a mean age (standard deviation (SD)) of 40.5 (14.2) years and a mean BMI (SD) of 23.5 (3.1) kg/m2. Twenty-two of the 24 subjects were white, one was Asian, and one was American Indian or Alaska Native.
A combined summary of study participants characteristics for each cohort is presented in Table 1.
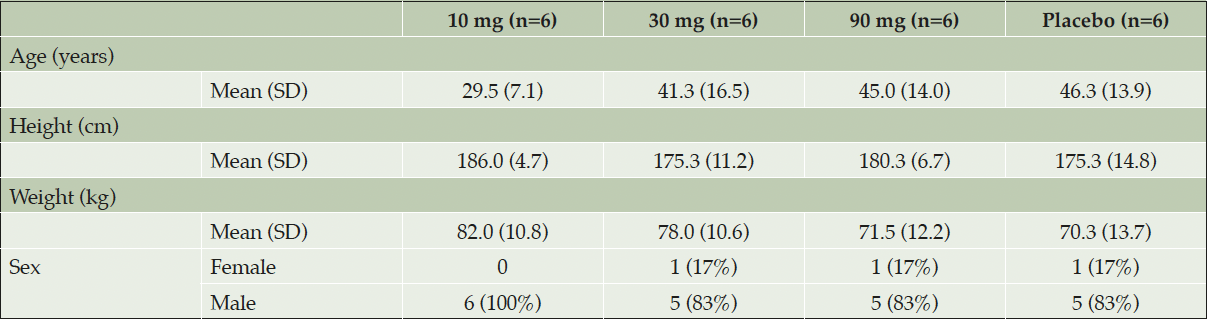
Table 1. Summary of study participant characteristics
Abbreviations: SD=standard deviation, n=total number of subjects
Safety and Tolerability
Treatment with ACD856 was generally well tolerated, without serious adverse events. No dose related trends were observed for AEs, laboratory safety assessments, vital signs, 12-lead ECG, physical examination, or stool. There was no indication of increased anxiety, depression, suicidal ideation, or suicidal behavior in any subject over the course of this study.
Twenty-two out of 24 subjects reported a total of 80 AEs, see supplementary Table S2. Most AEs (70 events reported by 22 subjects) were of mild intensity, the remaining 10 AEs reported by 7 subjects were assessed as moderate. In total, 12 events (reported by 10 subjects) were assessed as possibly related to study treatment and were reported by subjects receiving ACD856. Out of these, two events were reported as being of moderate intensity: a transient increase in lipase and amylase in one asymptomatic subject following the last day of ACD856 treatment. The most common AE was headache, which was reported by 12 subjects on 15 occasions, whereof 7 events were assessed as related to the lumbar puncture procedure for CSF sampling, and two were assessed as related to the study treatment (of which one was in a subject exposed to 90 mg ACD856). There was no indication of increasing AE reporting frequency with increasing dose of ACD856 and no obvious difference in the AE reporting between subjects receiving active treatment and subjects receiving placebo frequency.
The cardiac safety analysis concluded that ACD856 administered once daily for 7 days has no or only small effects on the cardiac conduction system of the heart. No systematic significant treatment induced change from baseline in heart rate or cardiac intervals was observed. The largest absolute QTcF value was 443 ms for one subject in the placebo group during the day of first dose. The largest individual change in QTcF (22 ms) was observed on Day 7 in a subject treated with ACD856 30 mg. The comparison of the mean changes from baseline in QTcF by dose group did not reveal any systematic dose-dependent prolongation or shortening of the cardiac de- and repolarization duration.
Pharmacokinetics of Multiple Doses of ACD856
Multiple dose plasma PK of ACD856 was evaluated following oral administration of 10, 30 and 90 mg once daily for 7 days. ACD856 was rapidly absorbed and reached maximum plasma concentrations 30 to 45 minutes after administration both on Day 1 and Day 7, (see Table 2 and Figure 2). The Ctrough concentrations suggested that the steady state conditions were established before Day 6.
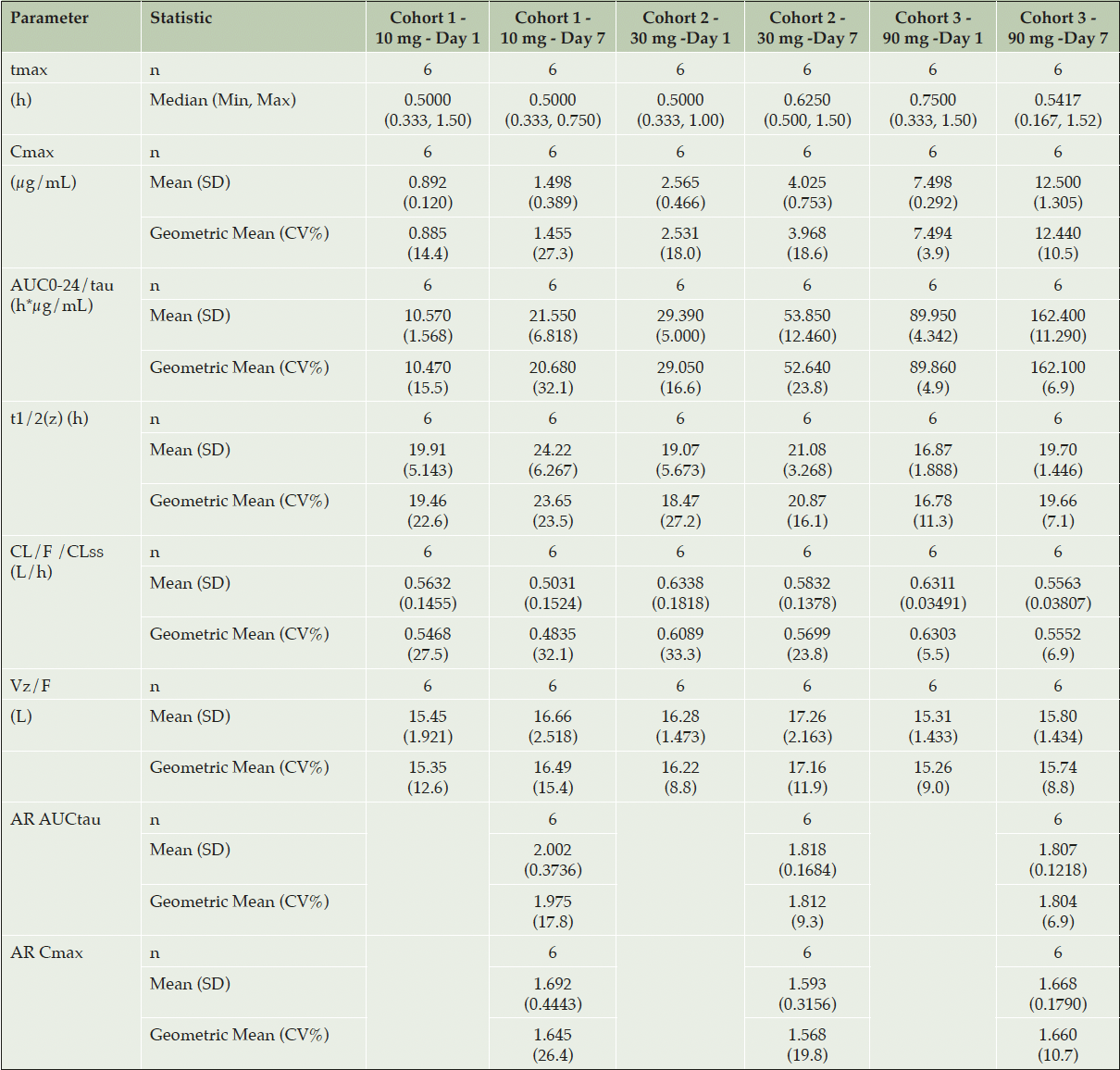
Table 2. Summary of Pharmacokinetic Parameters of ACD856 Following Repeated Oral Administration for 7 Days
Abbreviations: AR = Accumulation ratio; AUC = area under the plasma concentration-time curve; AUC0-24/tau = AUC over the dosing interval; CL/F/CLss = apparent oral clearance at steady state; Cmax = maximum observed plasma concentration; CV% = percent coefficient of variation; n = total number of subjects; t1/2(z) = terminal elimination half-life; tmax = time to Cmax; Vz/F = apparent volume of distribution associated with the terminal phase
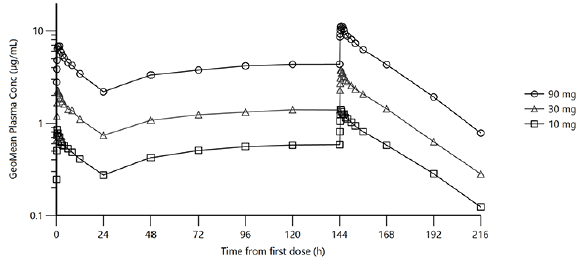
Figure 2. Plasma concentration curves for ACD856 throughout the multiple ascending dose study
Illustrates the geometric mean plasma concentration curves for ACD856 following repeated oral administration of 10 mg, 30 mg and 90 mg, once daily for 7 days in the multiple ascending dose study. The concentration is shown by time in hours from the first dose until the last measurement, 16 days after the first dose.
The exposure, both in terms of Cmax and AUC0-24, increased proportionally with dose on Day 1 within the dose range 10 to 90 mg. The dose proportionality constants at Day 1 for Cmax and AUC0-24 were 0.972 and 0.978, respectively. Although the dose increase appeared to be linear also at steady state on Day 7, this was not statistically confirmed. At steady state, the dose proportionality constants for Cmax and AUCtau were 0.977 and 0.937, respectively.
The plasma concentration of ACD856 accumulated following repeated dosing. At steady state on Day 7, the geometric mean accumulation ratios ranged between 1.8 and 2.0 for AUCtau and between 1.6 and 1.7 for Cmax. ACD856 exhibited a low volume of distribution (13.42-16.94 L) and a low clearance (0.48-0.63 L/h) with no obvious time dependency. The geometric mean elimination half-life at steady state ranged from 19.7-23.7 hours.
The inter-individual variation (CV%) was in general low, in particular in the highest dose group where the CV% values for all parameters (Day 1 and Day 7) ranged between 3.9% and 11.3%.
All collected urine samples exhibited ACD856 concentrations below the detection limit of the method (LLOQ=100 ng/mL) and hence the amount of ACD856 eliminated renally could not be calculated. Thus, renal elimination of ACD856 appears to be non-existing or negligible.
The MIST analysis result of the human plasma suggested that, in general, the levels of human metabolites were low in all dose cohorts with unchanged ACD856 constituting 77-86% of total drug related material detected. The metabolic profile in plasma and urine showed that ACD856 was extensively metabolized before being excreted in the urine. In total, 16 metabolites were detected and characterized in plasma based on their accurate masses, retention time and MS fragmentation, as oxidation products in various positions, and a combination of methylation, sulphate and glucuronic acid conjugation products. The apparent major metabolite (M1) constituted 6.5-11% of total ACD856 related material in human plasma, assuming equal MS response. Metabolite M3 constituted 2.5-2.9%, whereas all other metabolites were below 1.3% of total. In all human dose cohorts, the identified metabolites with levels above 1% of total drug related material have been previously identified and adequately exposed in rat and minipig 28-day toxicity studies. In the urine, a total of 17 metabolites were identified of which 13 were characterized. No ACD856 was excreted unchanged, and the metabolic pathways in vivo included oxidations followed by methylation, glucuronic acid or sulphate conjugation, similar to that seen in plasma.
The blood-brain barrier penetration of ACD856 was evaluated by analyzing the concentration of ACD856 in CSF on Day 6 where steady state conditions would likely have been established. The geometric mean concentration of ACD856 in CSF increased with dose: 3.98 ng/mL in the 10 mg dose cohort, 13.8 ng/mL in the 30 mg dose cohort, and 100 ng/mL in the 90 mg dose cohort. Prior to initiating this study, the plasma protein binding was assessed by in vitro equilibrium dialysis, resulting in an average estimated unbound fraction of 0.012 in human plasma. At steady state, the geometric mean ACD856 concentrations in CSF were similar to the estimated unbound average ACD856 plasma concentrations over the dosing interval (calculated geometric mean ratios ranged between 0.37 and 1.20). A summary of descriptive statistics for observed ACD856 CSF concentrations at steady state and the CSF/plasma concentration ratios (calculated by dividing the CSF concentration with the average estimated unbound plasma concentration over the dosing interval at steady state on Day 7) is shown in Table 3.
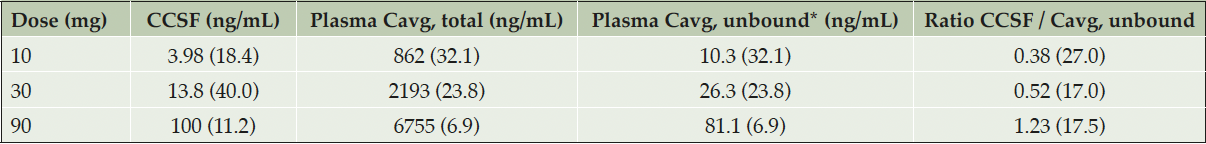
Table 3. ACD856 CSF and plasma concentrations at steady state (Day 6)
*Fu, plasma value used for estimation of unbound concentration was 1.2%; Abbreviations: Cavg = observed average plasma concentration; CCSF = concentration in cerebrospinal fluid. Values represent geometric mean (coefficient of variation in percent).
Quantitative EEG
Significant dose-dependent changes in qEEG parameters could be observed following ACD856 treatment, compared to pre-dose, whilst only spurious findings were observed after placebo treatment (refer to Figure 3 and supplementary Figures S1 and S2). The changes were most prominent for the 90 mg dose cohort. Dosing with ACD856 was statistically significantly associated with increased relative theta power and decreased fast alpha and beta power, and an increased theta/beta ratio (TBR). These changes were more robust when comparing intra-individual qEEG data (TBR: p<0.01) (Figure 3 and supplementary Figures S1 and S2) than when comparing the 90 mg group with the placebo group (TBR: p<0.05) (supplementary Figures S3 and S4), likely because of the small sample sizes. The changes were most evident in the vigilance-controlled EEG recordings with eyes closed, as opposed to resting EEG recordings with eyes closed or open (supplementary Figures S5 and S6) and were most pronounced at 24 hours post first dose of ACD856 and 1.5 hours after dosing on Day 7. Limited or no changes were observed at 1.5 and 6 hours post first dose versus pre-dose.
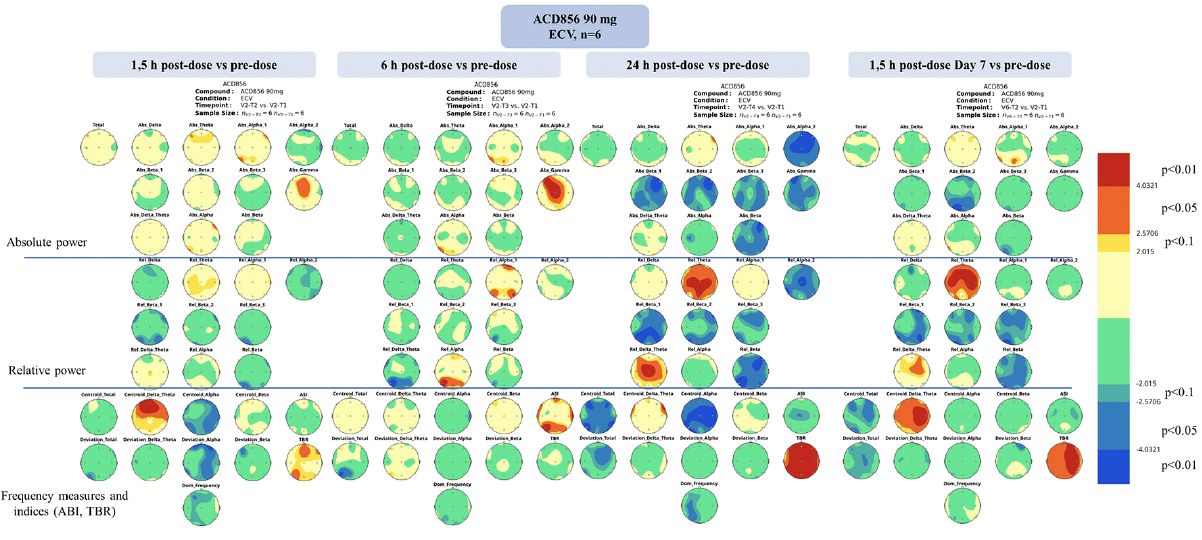
Figure 3. Statistical probability maps of qEEG results after 90 mg ACD856 dosing vs pre-dose at the analyzed time-points
Statistical probability maps of quantitative electroencephalography (qEEG) results comparing qEEG measurements after ACD856 90 mg treatment with pre-dose in the vigilance-controlled condition with eyes closed (ECV). The timing of the measurements was 1.5, 6 and 24 hours after the initial dose, as well as 1.5 hours after the last dose on day 7. The maps are based on paired samples t-tests and the number of subjects included was 6. The upper part shows the maps for the absolute power variables, the middle part the relative power variables and the lower part for the frequency measures as well as the alpha slow-wave index (ASI) and theta/beta ratio (TBR). Warm colors indicate treatment induced increases compared to pre-dose; cold colors indicate decreases. The thresholds are set according to the degree of freedom to significance levels at p<0.10, p<0.05 and p<0.01 (see legend on the right).
Discussion
Targeting the neurotrophin signaling pathways has been challenging due to their complex biology, limiting the attempts to develop therapeutics with this target (27). ACD856 exhibits a novel mechanism of action as a positive allosteric modulator of the Trk-receptors and has delivered promising preclinical data with regards to both pro-cognitive and antidepressant like effects (28). Furthermore, there is emerging preclinical in vitro and in vivo data suggesting that ACD856 may also have disease modifying effects, such as effects on long-term plasticity, neuroprotection, neurite outgrowth, mitochondrial function and cell membrane integrity in addition to previously reported symptomatic cognitive effects (29).
The first steps have now been taken towards better understanding the clinical potential for this novel mechanism of action. Preceding microdose and SAD studies indicated that ACD856 as single doses (0.1-150 mg) in healthy subjects was well tolerated and had a suitable PK profile for further development.
No treatment emergent or dose related trends were observed for any of the safety parameters following 7 days of treatment with ACD856 in healthy subjects. In one subject, a transient single timepoint and asymptomatic increase of lipase and amylase was detected following the last day of treatment, was assessed by the investigator as possibly related to the study drug in a lack of other explanations. Values returned to normal within 24 hours. The MAD study safety data is in line with results seen in the ACD856 single dose clinical studies, as well as the preclinical toxicology studies which showed no safety findings.
Similar to the observed single dose data (30), multiple doses of ACD856 suggest a pharmacokinetic profile suitable for further clinical development with a rapid absorption and a dose-dependent increase in exposure. The inability to detect unchanged ACD856 in urine indicates that, as is often seen for highly lipophilic compounds, ACD856 appears to be metabolized through oxidation, and subsequent methylation, glucuronidation and sulfation pathways before being excreted in the urine. The metabolite profiling raises no concerns with regard to toxicity caused by metabolites as the pattern is similar to the metabolites observed in animal studies.
Measured levels of ACD856 in CSF confirm that ACD856 crosses the blood-brain-barrier and adds confidence that relevant levels are reaching the target receptors in the brain. The exposure in CSF increased with increasing doses of ACD856 in the dose range 10 to 90 mg (determined at steady state). The measured levels of ACD856 in CSF at the highest dose level (geometric mean concentration 100 ng/mL at steady state for 90 mg/day dosing), assuming no or limited protein binding in the CSF, is well above (>20 times) the estimated unbound brain exposure measured at effective doses of ACD856 in mice showing a pro-cognitive effect (28). However, considering the inherent uncertainty in translatability from preclinical observations to clinical predictions, conclusions regarding the potential clinical effective dose should be drawn with caution and need confirmation by clinical studies in the relevant target patient population.
ACD856 treatment demonstrated dose-dependent treatment-associated changes on qEEG parameters in healthy subjects. This is a promising observation given that electrophysiological methods such as resting EEG have been proposed as BDNF sensitive functional markers in early clinical development to examine target engagement (25). Specifically, ACD856 treatment was statistically significant associated with increased relative theta power and decreased fast alpha and beta power in a dose-dependent fashion which led to an acceleration of the delta+theta centroid and an increase in the theta/beta ratio. These findings are in line with EEG effects reported for BDNF knock-out rats showing that a reduction in BDNF content is related to a decrease in hippocampal delta and theta activity (47). In addition, a study in patients with gambling disorder showed a positive correlation between serum BDNF levels and theta EEG power in the right parietal region (48). The treatment-associated changes seen on qEEG parameters following ACD856 treatment in healthy subjects are, together with the evidence of dose-dependent increase of measured ACD856 levels in CSF, an indication of target engagement. This may mean that ACD856 is able to exert its hypothesized mode of action in humans, i.e., increased neurotrophin signaling.
The ACD856 EEG result pattern in healthy subjects is similar to what is seen during the physiological state of relaxation (49). It is also similar to the EEG profile reported for buspirone (a partial serotonin receptor 1A (5-HT1A) agonist and dopamine D2/D3 receptor antagonist) in healthy subjects (49, 50). Buspirone is approved for the long-term treatment of anxiety and restlessness. Looking at donepezil hydrochloride, an acetylcholinesterase inhibitor used for the treatment of AD, a study in older healthy volunteers showed that impaired memory was associated with a decrease in EEG alpha and beta power and an increase in delta power (51). In contrast, after six months of treatment with donepezil in patients with AD dementia, cognition (Mini Mental State Examination) was improved, EEG delta activity decreased, and alpha and beta activity increased (52). Any cross-reference to studies exploring other CNS active drugs should only be done with caution, considering differences in study populations and in EEG methodology. Nevertheless, it may be of interest to better understand whether ACD856 exerts any effects on qEEG parameters in a relevant patient population and if this differs from the activity patterns seen in healthy subjects.
The main inherent limitation of the current phase I study is the small study population. The number of subjects per dose cohort is in agreement with available guidance for FIH ascending dose studies (38) and are deemed sufficient to evaluate safety in healthy subjects in this early type of phase I trial. However, extensive data on the safety of ACD856 treatment in humans remains to be collected in clinical trials with larger sample sizes and longer treatment duration in order to fully understand the safety profile of ACD856. The limited sample size should also be remembered in the review of the pharmacokinetic and pharmacodynamic results (i.e., qEEG parameters). Pharmacokinetic results should be further assessed in larger clinical trials in order to determine if the highly predictable PK properties can be confirmed. The observed dose-dependent treatment-associated changes on qEEG parameters, while statistically significant, do not allow for any general conclusions regarding effects of ACD856 treatment on neuronal activity. Furthermore, the level of relaxation or vigilance in the study subjects during the qEEG measurement was not systematically evaluated and no active control treatments with known qEEG patterns were used.
In conclusion, ACD856 was well tolerated at the tested multiple dose levels (10-90 mg/daily for 7 days) in healthy subjects. No safety signals were observed. PK results indicate that ACD856 has a suitable pharmacokinetic profile for further development with a rapid absorption, dose-dependent exposure, and a favorable metabolic profile. The CSF concentration results show that ACD856 passes the blood-brain-barrier and reaches relevant exposure in the brain as these are well above the observed brain exposure in preclinical studies for which pro-cognitive effects of ACD856 were demonstrated (27, 28). Furthermore, qEEG data indicates that ACD856 not only passes the blood brain barrier but also exerts some treatment-associated changes on CNS activity, which suggests that central target engagement is achieved. The clinical relevance of this finding remains to be understood and is to be evaluated in a relevant patient population. The next step in the clinical development of ACD856 will be to evaluate its effects in a relevant patient population to understand if ACD856 could prove to be an effective treatment option for patients with AD, depression, other psychiatric disorders and disorders where cognition is impaired.
Acknowledgments: The authors wish to thank the study participants at the clinical phase I unit in Uppsala, Sweden. We also gratefully acknowledge the investigators, study coordinators and the contract research organization CTC Clinical Trial Consultants AB for their support with the studies. Cardiac safety was well arranged and analyzed by Nabios GmbH. We want to thank Lablytica Life Science AB for analysis of ACD856 in plasma, urine, and CSF, and MetaSafe Sweden AB for the metabolic evaluation. In addition, we want to acknowledge RISE Research Institutes of Sweden AB for their support with development and production of the investigational medicinal product.
Conflict of interest: KÖ, BN, JS and MS are employees of AlzeCure Pharma. MR and MH are paid consultants of AlzeCure Pharma. ER and JB are employees of CTC Clinical Trial Consultants AB. PA is a paid consultant of The Siesta Group Schlafanalyse GmbH and an employee of Philips Austria GmbH, and MK is employed by The Siesta Group Schlafanalyse GmbH. CTC Clinical Trial Consultants AB and The Siesta Group Schlafanalyse GmbH are contract services providers for AlzeCure Pharma.
Ethical standards: The current clinical study was conducted in compliance with the study protocol, the International Council for Harmonization Guideline for Good Clinical Practice, all applicable regulatory requirements, and in accordance with the latest revision of the Ethical Principles for Medical Research Involving Human Subjects (the Declaration of Helsinki). The study protocol and all applicable study-related documentation were approved by the Swedish Ethical Review Authority and the Swedish Medical Products Agency. Signed informed consent was obtained from the study participants prior to any study-related activities. The study was conducted at the First-in-Human unit of CTC Clinical Trial Consultants AB, Uppsala, Sweden.
Funding: The sponsor, AlzeCure Pharma AB, provided full financial support for the current clinical studies and associated activities. The sponsor was involved in study design, data collection, analysis, data interpretation, and preparation of the manuscript. Open Access funding provided by Karolinska Institute.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Alzheimer’s Association. 2022. Alzheimer’s Disease Facts and Figures. Alzheimers Dement 2022;18. https://doi.org/10.1002/alz.12638
2. Cummings J, Lee G, Nahed P, et al. Alzheimer’s disease drug development pipeline: 2022. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2022;Volume 8, Issue 1. https://doi.org/10.1002/trc2.12295
3. Tricco AC, Soobiah C, Berliner B, et al. Efficacy and safety of cognitive enhancers for patients with mild cognitive impairment: a systematic review and meta-analysis. CMAJ 2013;185(16); 1393-1401. https://doi.org/10.1503/cmaj.130451
4. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in Early Alzheimer’s Disease. The New England Journal of Medicine. 2022;November 29, 2022. https://doi.org/10.1056/NEJMoa2212948
5. Budd Haeberlein S, Aisen P, Barkhof F, et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. The journal of prevention of Alzheimer’s Disease, 2022;9, 18 March 2022. https://doi.org/10.14283/jpad.2022.30
6. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in Early Alzheimer’s Disease. The New England Journal of Medicine. May 6, 2021. https://doi.org/ 10.1056/NEJMoa2100708
7. Chao MV. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nature Reviews Neuroscience 2003;4; 299-309. https://doi.org/10.1038/nrn1078
8. Leal G, Afonso PM, Salazar IL, and Duarte CB. Regulation of hippocampal synaptic plasticity by BDNF. Brain Research 2015;1621; 82-101. https://doi.org/10.1016/j.brainres.2014.10.019
9. Counts SE, Mufson EJ. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J Neuropathology Exp Neurology 2005;64; 263–272. https://doi.org/10.1093/jnen/64.4.263
10. Peng S, Wuu J, Mufson EJ, et al. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease, J Neuropathology Exp Neurology 2004;63; 641–649. https://doi.org/10.1093/jnen/63.6.641
11. Peng S, Wuu J, Mufson EJ, et al. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J Neurochem. 2005;93(6):1412-21. doi: 10.1111/j.1471-4159.2005.03135.x.
12. Jiao SS, Shen LL, Zhu C, et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Translational Psychiatry 2016;6(10); e907. https://doi.org/10.1038/tp.2016.186
13. Kemppainen S, Rantamäki T, Jerónimo-Santos A, et al. Impaired TrkB receptor signalling contributes to memory impairment in APP/PS1 mice. Neurobiology of Aging 2012;33(6); 1122.e23-29. https://doi.org/10.1016/j.neurobiolaging.2011.11.006
14. Nagahara AH, Merrill DA, Coppola G, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nature Medicine 2009;15(3); 331-337. https://doi.org/10.1038/nm.1912
15. Arancibia S, Silhol M, Moulière F, et al. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats, Neurobiol Dis; 2008;316–326. https://doi.org/10.1016/j.nbd.2008.05.012
16. Jiao SS, Shen LL, Zhu C, et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease, Transl Psychiat 2016;6; e907. https://doi.org/10.1038/tp.2016.186
17. Neto FL, Borges G, Torres.Sanchez, et al. Neurotrophins Role in Depression Neurobiology: A Review of Basic and Clinical Evidence. Current Neuropharmacology, 2011; Dec; 9(4): 530–552. https://doi.org/ 10.2174/157015911798376262
18. Casarotto PC, Giryck M, Fred SM, et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell. 2021; Mar 4. https://doi.org/ 10.1016/j.cell.2021.01.034
19. Egan MF, Kojima M, Callicott, JH, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. Volume 2003;112. https://doi.org/10.1016/s0092-8674(03)00035-7
20. Kambeitz, JP, Bhattacharyya S, Kambeitz-Ilankovic, LM, et al. Effect of BDNF val(66)met polymorphism on declarative memory and its neural substrate: a metaanalysis. Neuroscience & Biobehavioral Reviews. 2012;Vol 36. https://doi.org/10.1016/j.neubiorev.2012.07.002
21. Miranda M, Morici JF, Zanoni MB, et al. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Frontiers in Cellular Neuroscience. Published 2019; Aug 7. http://doi.org\10.3389/fncel.2019.00363
22. Wang Q, Liu J, Guo Y, et al. Association between BDNF G196A (Val66Met) polymorphism and cognitive impairment in patients with Parkinson’s disease: a meta-analysis. Brazilian Journal of Medical and Biological Research, 2019;52(8). https://doi.org/10.1590/1414-431X20198443
23. Flores KR, Viccaro F, Aquilini M, et al. Protective role of brain derived neurotrophic factor (BDNF) in obstructive sleep apnea syndrome (OSAS) patients. PLoS One. 2020;15(1): e0227834. https://doi.org/10.1371/journal.pone.0227834
24. Bachmann V, Klein C, Bodenmann S, et al. The BDNF Val66Met polymorphism modulates sleep intensity: EEG frequency- and state-specificity. Sleep, 2012;Mar 1; 35(3). https://doi.org/10.5665/sleep.1690
25. Soltész F, Suckling J, Lawrence P, et al. Identification of BDNF Sensitive Electrophysiological Markers of Synaptic Activity and Their Structural Correlates in Healthy Subjects Using a Genetic Approach Utilizing the Functional BDNF Val66Met Polymorphism. PLoS One. 2014; 9(4). https://doi.org/10.1371/journal.pone.0095558
26. Roy N, Barry RJ, Fernandez FE, et al. Electrophysiological correlates of the brain-derived neurotrophic factor (BDNF) Val66Met polymorphism. Scientific Reports, 2020;Oct 21;10(1). https://doi.org/10.1038/s41598-020-74780-9
27. Nordvall G, Forsell P, Sandin S. Neurotrophin-targeted therapeutics: A gateway to cognition and more? Drug Discovery Today 2022;Volume 27; Number 10. https://doi.org/10.1016/j.drudis.2022.07.003
28. Dahlström M, Madjid N, Nordvall G, et al. Identification of Novel Positive Allosteric Modulators of Neurotrophin Receptors for the Treatment of Cognitive Dysfunction. Cells 2021, 10, 1871. https://doi.org/10.3390/cells10081871
29. Parrado-Fernández C, Nordvall G, Juric S, et al. Preclinical characterization of ACD856, a cognitive enhancer in clinical development for the treatment of cognitive dysfunction in Alzheimer’s disease, demonstrates increased plasticity, neuroprotection and a possible disease modifying effect. Journal of Prevention of Alzheimer’s Disease-JPAD, Volume 2022;9, Suppl. 1, S84-85. https://www.ctad-alzheimer.com/files/files/JPAD%20CTAD%20abstracts.pdf
30. Nilsson B, Rother M, Rein-Hedin E, et al. 2023. ACD856, a Novel Positive Allosteric Modulator of Trk-receptors, Single Ascending Doses in Healthy Subjects: Safety and Pharmacokinetics. EJCP, submitted.
31. Apfel SC, Schwartz S, Adornato BT, et al. Efficacy and Safety of Recombinant Human Nerve Growth Factor in Patients With Diabetic Polyneuropathy, A Randomized Controlled Trial. JAMA, 2000;284:2215-2221. https://doi.org/10.1001/jama.284.17.2215
32. Coulie B, Szarka LA, Camilleri M, et al. Recombinant human neurotrophic factors accelerate colonic transit and relieve constipation in humans. Gastroenterology. 119(1):41-50. https://doi.org/10.1053/gast.2000.8553
33. Drilon A. TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019 Nov 1;30(Suppl_8): viii23-viii30. https://doi.org/10.1093/annonc/mdz282
34. Eriksdotter Jönhagen M, Nordberg A, Amberla K, et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 1998;9(5):246-57. https://doi.org/10.1159/000017069
35. Hong DS, DuBois SG, Kummar S, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020 Apr;21(4):531-540. https://doi.org/10.1016/S1470-2045(19)30856-3.
36. Sahenk Z, Nagaraja HN, McCracken BS, et al. NT-3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology. 2005 Sep 13;65(5):681-9. https://doi.org/10.1212/01.wnl.0000171978.70849.c5.
37. A controlled trial of recombinant methionyl human BDNF in ALS: The BDNF Study Group (Phase III). (1999) Neurology. 1999;22;52(7):1427-33. https://doi.org/10.1212/wnl.52.7.1427.
38. Committee for Medicinal Products for Human Use. Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. European Medicines Agency. 20 July 2017. EMEA/CHMP/SWP/28367/07 Rev. 1.
39. National Cancer Institute Division of Cancer Treatment and Diagnosis, Cancer Therapy Evaluation Program, 2017. Common terminology criteria for adverse events. CTCAE v5.0. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm. (Accessed 26 January 2023).
40. Spitzer RL, Kroenke K, Williams JBW, Löwe B. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166(10). https://doi.org/ 10.1001/archinte.166.10.1092
41. Kroenke K, Spitzer RL, Williams JBW. The PHQ-9. Journal of General Internal Medicine, 2001;16. https://doi.org/10.1046/j.1525-1497.2001.016009606.x
42. Posner K, Brown GK, Stanley B, et al. The Columbia–Suicide Severity Rating Scale: Initial Validity and Internal Consistency Findings From Three Multisite Studies With Adolescents and Adults. Published online 1 Dec 2011. https://doi.org/10.1176/appi.ajp.2011.10111704
43. European Medicines Agency Committee for Medicinal Products for Human Use (CHMP), 2011. Guideline on bioanalytical method validation, EMEA/CHMP/EWP/192217/2009. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (last accessed 31May2023)
44. European Medicines Agency Committee. ICH M3 (R2) Non-clinical safety studies for the conduct of human clinical trials for pharmaceuticals – Scientific guideline, Step 5 December 1 2009 (ema.europa.eu) (last accessed 31May2023)
45. US Food and Drug Administration, 2020. Guidance for Industry: Metabolites in Safety Testing. Safety Testing of Drug Metabolites Guidance for Industry (fda.gov) (last accessed 31May2023)
46. Jobert M, Wilson FJ, Ruigt GSF, et al. Guidelines for the recording and evaluation of pharmaco-EEG data in man: The International Pharmaco-EEG Society (IPEG). Neuropsychobiology 2012;66(4): 201-220. https://doi.org/10.1159/000343478
47. Geist PA, Dulka BN, Barnes A, et al. BNDF heterozygosity is associated with memory deficits and alterations in cortical and hippocampal EEG power. Behavioural Brain Research, 2017;Vol 332. https://doi.org/10.1016/j.bbr.2017.05.039
48. Kim KM, Choi S-W, Lee J, Kim JW. EEG correlates associated with the severity of gambling disorder and serum BDNF levels in patients with gambling disorder. Journal of Behavioral Addictions. 2018;Jun; 7(2). https://doi.org/10.1556/2006.7.2018.43
49. Anderer P, Saletu B, Pascual-Marqui RD, 2000. Effect of the 5-HT(1A) partial agonist buspirone on regional brain electrical activity in man: a functional neuroimaging study using low-resolution electromagnetic tomography (LORETA). Psychiatry Research: Neuroimaging Section 100. https://doi.org/10.1016/s0925-4927(00)00066-4
50. Barbanoj MJ, Anderer P, Antonijoan RM, et al. Topographic pharmaco-EEG mapping of increasing doses of buspirone and its comparison with diazepam. Human Psychopharmacology. 1994; Volume 9. https://doi.org/10.1002/hup.470090203
51. Balsters JH, O’Connell RG, Martin MP, et al. Donepezil impairs memory in healthy older subjects: behavioural, EEG and simultaneous EEG/fMRI biomarkers. PLoS One. 2011; 6(9): e24126. https://doi.org/10.1371/journal.pone.0024126
52. Balkan S, Yaraş N, Mihçi E, et al. Effect of donepezil on EEG spectral analysis in Alzheimer’s disease. Acta Neurologica Belgica 2003;103
© The Authors 2023