
E. Trushina1,2, T.K.O. Nguyen1, S. Trushin1
1. Department of Neurology, Mayo Clinic, Rochester, Minnesota, USA; 2. Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, Minnesota, USA
Corresponding Author: Eugenia Trushina, Ph.D., Mayo Clinic, 200 First Street SW, Guggenheim Building 1442B, Rochester, MN 55905, USA, Phone: 507-284-8197, trushina.eugenia@mayo.edu
J Prev Alz Dis 2023;4(10):675-685
Published online October 24, 2023, http://dx.doi.org/10.14283/jpad.2023.108
Abstract
Despite recent FDA approval of anti-amyloid antibodies for Alzheimer’s Disease, strategies that target early molecular mechanisms and could delay or change the disease trajectory are still needed. Mitochondria emerge as a signaling organelle that could modulate multiple molecular mechanisms to enhance cellular bioenergetics and promote neuronal survival. Approaches to enhance mitochondrial function could promote healthy aging delaying the onset of age-related diseases such as Alzheimer’s Disease. Some of these strategies have been recently tested in clinical trials. Emerging evidence demonstrates that in response to mild energetic stress, mitochondria could orchestrate a robust adaptive stress response activating multiple neuroprotective mechanism. The objective of this review is to highlight recent development of mitochondria-targeting therapeutics for neurodegenerative diseases, mitochondria complex I inhibitors in particular.
Key words: Mitochondria, mitochondrial signaling, complex I inhibitors, neurodegenerative diseases of aging, Alzheimer’s Disease, neuroprotection, mitochondria targeting therapeutics.
Introduction
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder where mechanistic changes that lead to amyloid beta (Aβ) and hyper‒phosphorylated Tau protein (pTau) accumulation, the AD hallmarks, may start ~20 years prior to the onset of cognitive changes (1). It is now increasingly recognized that early AD mechanisms include metabolic dysfunction manifested in abnormal glucose/energy metabolism and increased neuronal damage (2). Synaptic degeneration is the best neuropathological correlate of cognitive decline in AD (3). While brain hypometabolism has no single cause, contributing mechanisms converge on alterations in mitochondrial dynamics and function, increased calcium levels and oxidative stress that affect cellular energy homeostasis. Neurons are most likely to be affected in AD under conditions of reduced glucose uptake, one of the earliest abnormalities detected the brain of people with prodromal AD, since the maintenance of synaptic function relies heavily on a consistent supply of energy. This energy is produced in mitochondria via a process known as oxidative phosphorylation (OXPHOS) with glucose as a major fuel for this process. Mitochondria are abundant in brain cells occupying ~25% of cell volume. The mitochondrion is the only cellular organelle other than the nucleus that has its own DNA (mtDNA) and transcriptional and translational machinery. These features, together with the unusual dynamics where organelles divide and fuse depending on their life cycle or in response to energy fluctuations, have supported the theory of an ancient endosymbiosis of a nucleated cell and an aerobic prokaryote (4). Benefits of this cooperation include mitochondria becoming the “power plant” of the cell, which significantly increased energy supply, while mitochondria outsourced the most of protein synthesis to the host cell. As a result, a robust communication system has been developed between mitochondria and the nucleus, known as mito-nuclear communication, and between mitochondria and other subcellular compartments and organelles (5). Thus, it is not surprising that apart from the generation of adenosine triphosphate (ATP), mitochondria acquired key roles in maintaining cellular bioenergetics, genomic stability, redox state, ion homeostasis, lipid and heme biosynthesis, innate immune surveillance, active stress responses, ultimately determining the fate of the cell (6, 7). Below, we highlight the current understanding of the role mitochondria play in health and disease and the development of mitochondria-targeted therapeutics for neurodegenerative diseases of aging (NDA).
Mitochondrial signaling in health and disease
The fidelity of the OXPHOS machinery is essential to support neuronal activity (8). ATP is produced in mitochondria during a series of metabolic reactions that include oxidation of glucose or its metabolites producing ~ 36 ATP molecules (9) (Fig. 1). During the OXPHOS, four protein complexes of the electron transport chain (ETC) move electrons received from nicotinamide adenine dinucleotide (NADH) or flavin adenine dinucleotide (FADH2) produced in the tricarboxylic acid (TCA) cycle via a series of redox reactions coupled to a final phosphorylation of adenosine diphosphate (ADP) to produce ATP, CO2, and water. The ETC (Fig. 1) includes four complexes: NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), cytochrome c oxidoreductase (complex III), and cytochrome c oxidase (complex IV) (10). Electrons moving through the ETC promote the translocation of protons (H+) from the mitochondrial matrix to the intermembrane space, establishing an electrochemical gradient or proton‒motive force (PMF). The energy generated from the PMF is used to phosphorylate ADP to ATP via ATP synthase (complex V). This process also includes the formation of reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) and superoxide (O2-) primarily by complexes l and lll. Under normal conditions, ROS generated during the OXPHOS are sequestered by the antioxidant enzymes such as superoxide dismutases, glutathione peroxidase, glutaredoxins, thioredoxins and catalases (11, 12). Under disease conditions or periods of high energy demand, the excessive ROS production may overwhelm the antioxidant capacities of the cell resulting in oxidative damage. Thus, mitochondrial function is tightly linked to the levels of ROS, energy metabolites, metabolites of the TCA cycle, and mitochondrial membrane potential (Δψm). With age or during the AD development, mitochondria show reduced capacity to produce ATP and maintain bioenergetics, they become more stationary within brain cells showing diminished ability to travel along the axons (anterograde and retrograde trafficking) to support axonal growth and synaptic function. Significant alterations were reported regarding the maintenance of a healthy mitochondrial pool through the process of biogenesis (a production of new organelles), mitophagy (the removal of damaged organelles), fission and fusion (changes in shape essential for quality control), which collectively increase mitochondrial ROS (11, 13-15). Mitochondria-produced ROS could damage mtDNA further accelerating mitochondrial dysfunction. The loss of mitochondrial function is considered a driving force behind the senescence, one of the prominent hallmarks of aging and the greatest risk factor for NDA (16). While mitochondria could influence multiple cellular mechanisms, it has been shown that the capacity of mitochondria to generate energy and sustain stress (mitochondrial fitness) alone could determine the survival of brain cells emphasizing the essential role mitochondria play in health and disease (15, 17, 18).
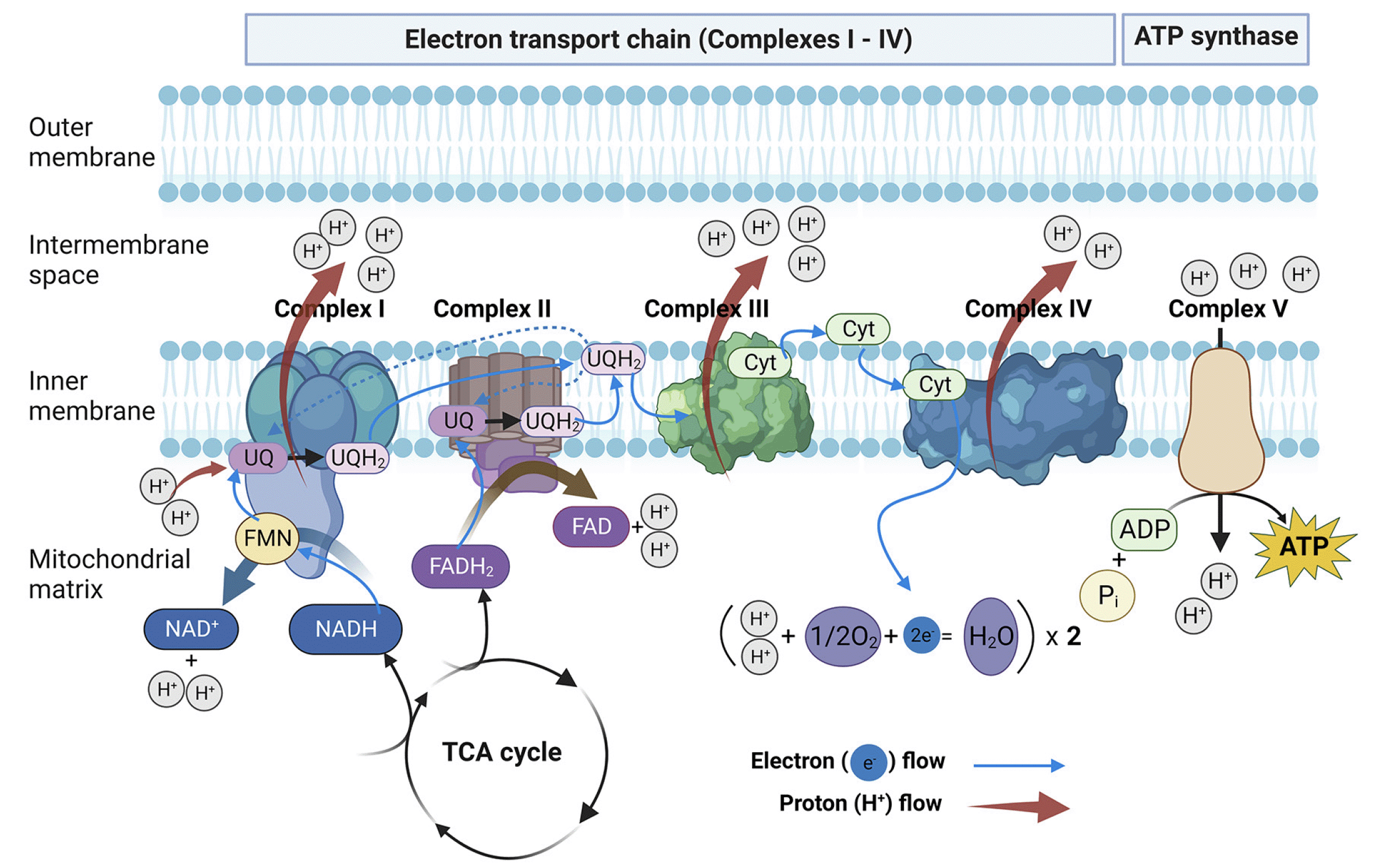
Figure 1. Schematic model of the oxidative phosphorylation (OXPHOS) process in mitochondria
Adenosine triphosphate (ATP) is generated by OXPHOS conducted by the electron transport chain (ETC) (Complexes I – IV) and ATP synthase (complex V) located in the inner mitochondrial membrane. Nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) are converted to nicotinamide adenine dinucleotide-NAD+ (complex I) and FAD (complex II), respectively, with H2O formed as a byproduct. Complex I then transfers both electrons to ubiquinone (UQ), forming its high energy form, UQH2. Like complex I, complex II receives two electrons from FADH2, oxidizing the molecule to its low energy form, FAD. However, unlike complex I, complex II does not pump protons across the inner membrane. Complex III, also known as cytochrome reductase, transfers electrons from ubiquinone to cytochrome C (Cyt c). Complex IV, also known as cytochrome oxidase, then transfers electrons from Cyt c to oxygen, producing water as a byproduct. Energy released by the ETC is used to force protons (H+) to flow from the mitochondrial matrix into the intermembrane space. The proton gradient across the inner mitochondrial membrane drives ATP production via ATP synthase to catalyze the generation of ATP from adenosine diphosphate (ADP) and inorganic phosphate (Pi). Figure was created with BioRender.com.
As mentioned above, mitochondria maintain cellular energy homeostasis and cell survival through a robust signaling system. The examples of mitochondrial intracellular communication include activation of mitochondrial biogenesis via engaging a co-transcriptional regulation factor peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) through metabolic sensor AMP-activated protein kinase (AMPK) in response to changes in ATP and AMP levels (19). Activation of PGC1α has been shown to elicit neuroprotective effects in models of Parkinson’s (PD) and AD (20, 21). Mitophagy, a process of removal of damaged organelles, could be induced by changes in mitochondrial Δψm, levels of NAD+, or activation of AMPK in PTEN-induced kinase 1 (PINK1) and E3 ubiquitin ligase Parkin dependent or independent manner (22-24). Both mitochondrial serine/threonine-protein kinase PINK1 and Parkin are neuroprotective proteins that act together to ensure the removal of damaged mitochondria (25). Mitophagy is balanced with biogenesis since the uncontrolled removal of mitochondria could result in abnormal energy homeostasis (26). Activation of mitophagy blocks neurodegeneration of glutamatergic and cholinergic neurons, protects against Aβ and pTau toxicity, and improves cognitive function in mouse models of AD (27).
The metabolites produced or controlled by mitochondria, including acetyl coenzyme A (acetyl-CoA), α-ketoglutarate, NAD+ and ROS, can regulate nuclear gene transcription activating antioxidant protection, mitigating inflammation, and inducing epigenetic modifications (28-30). The mitochondria-derived peptide mitochondrial open-reading-frame of the 12S rRNA‒C (MOTS-c) has been shown to translocate to the nucleus and regulate metabolic homeostasis via AMPK-dependent mechanism protecting against age- and diet-dependent insulin resistance and obesity, factors that contribute to the development of AD (31). Mitochondria regulate immune response by releasing mtDNA and through mitochondria-derived peptides (e.g., humanin and MOTS‒c) (32-34). Mitochondria-derived vesicles (MDVs) represent another critical element of mitochondrial quality control. MDVs carry damaged or oxidized components of organelles delivering them to lysosomes for degradation or releasing outside the cell via extracellular vesicles (EVs) (35). Under normal conditions, MDVs were linked to mitochondrial fission, peroxisome biogenesis, a resistance to oxidative stress, anti-infection, and boosting innate immunity while abnormal MDVs were implicated in neurodegenerative diseases promoting neuroinflammation (35-37). The interaction of mitochondria with the endoplasmic reticulum (ER), mito-ER communication, or Golgi has been shown to affect Ca2+ flux, lipid metabolism, autophagosome formation, ER stress, mitochondrial quality control, bioenergetics, apoptosis, and cellular architecture (38, 39). These examples highlight the complexity of mitochondrial signaling and the essential role of mitochondrial homeostasis in cellular functions.
In recent years, additional evidence emphasized the upstream role of mitochondria in the mechanisms of AD etiology. Changes in mitochondrial function have been shown to affect processing of Amyloid Precursor Protein (APP) and Aβ production (40), expression of apolipoprotein E (APOE) (41), and pTau aggregation (42). AD brains exhibit reduced expression of mitochondrial genes, mtDNA copy number, and low levels of proteins associated with mitochondrial respiratory chain involved in ATP production (43-46). In addition, it has been shown that mitochondria-mediated changes in energy homeostasis and signaling molecules involved in protective stress response, ROS generation, epigenetic modifications, neuroinflammation, and calcium homeostasis could independently determine the course of the disease (11, 47-49). The “mitochondrial cascade hypothesis” proposes that an individual’s genetic predisposition, environmental exposure, and lifestyle could affect mitochondrial function and mediate, drive, and/or contribute to a variety of AD pathologies (50). These mitochondrial changes are not restricted to the brain and could be detected in the periphery including plasma, lymphocytes, and fibroblasts (51-53). However, despite strong evidence implicating mitochondria in AD development, their diverse morphology, highly adaptive nature, and broad spectrum of functions convolute a clear mechanistic understanding of mitochondrial contribution to AD pathogenesis. Thus, determining the timing of interventions in the disease process and the optimal type of interventions targeting mitochondria has been challenging.
Mitochondria as a therapeutic target
While mitochondrial dysfunction is well documented in NDA, recent data also support mitochondria as a therapeutic target (54-57). In this section, we will focus on the development of mitochondria-targeted strategies for AD. Additional information regarding other neurovegetative diseases could be found in these outstanding reviews (54, 58, 59). Few pharmacological approaches to enhance mitochondrial biogenesis and mitophagy, to reduce oxidative stress, and promote glucose metabolism have been tested in clinical trials. Table 1 presents the examples of therapeutic strategies developed toward specific mitochondria mechanisms and representative clinical trials that tested these approaches in AD patients (55, 60-66). Comprehensive information on all past, ongoing, and recruiting clinical trials for AD can be found at Clinicaltrials.gov web site. While pharmacological strategies produced mixed results, intriguingly, the accumulating evidence suggests that non-pharmacological approaches, such as diet, exercise and mild mitochondrial stress induced by inhibition of the ETC components could reduce major AD hallmarks by engaging an adaptive stress response that leads to improved metabolic state, reduced oxidative stress and inflammation, and improved proteostasis (67-79). Over 70 clinical trials are testing efficacy of lifestyle interventions, including diet and exercise, on the AD trajectory (Table 1). Relevant to mitochondria, the longitudinal RNA sequencing (RNA-seq) analysis identified one of the major complexes in the OXPHOS machinery, mitochondrial complex I, as a hub in a module of genes whose expression was negatively correlated with lifespan in African turquoise killifish (74). Mild mitochondrial injury associated with the knockdown of various ETC complexes in D. melanogaster has been shown to increase longevity via hormetic signaling (80, 81). Of key importance to the extension of health span in humans, correlation between mtDNA variability and longevity in a cohort of 2200 centenarians revealed that mutations in subunits of complex I that resulted in partial loss of its activity increased longevity (76). While inhibition of complex I to achieve healthy aging and prevent NDA appears counterintuitive, broad application of mild complex I inhibitor metformin (82), an FDA-approved drug for type 2 diabetes, supports the feasibility and safety of such an approach in humans (83-89). Beneficial mechanisms associated with these strategies appear to converge on the induction of a mild energetic stress, activation of mitochondrial signaling and the adaptive stress response (90).
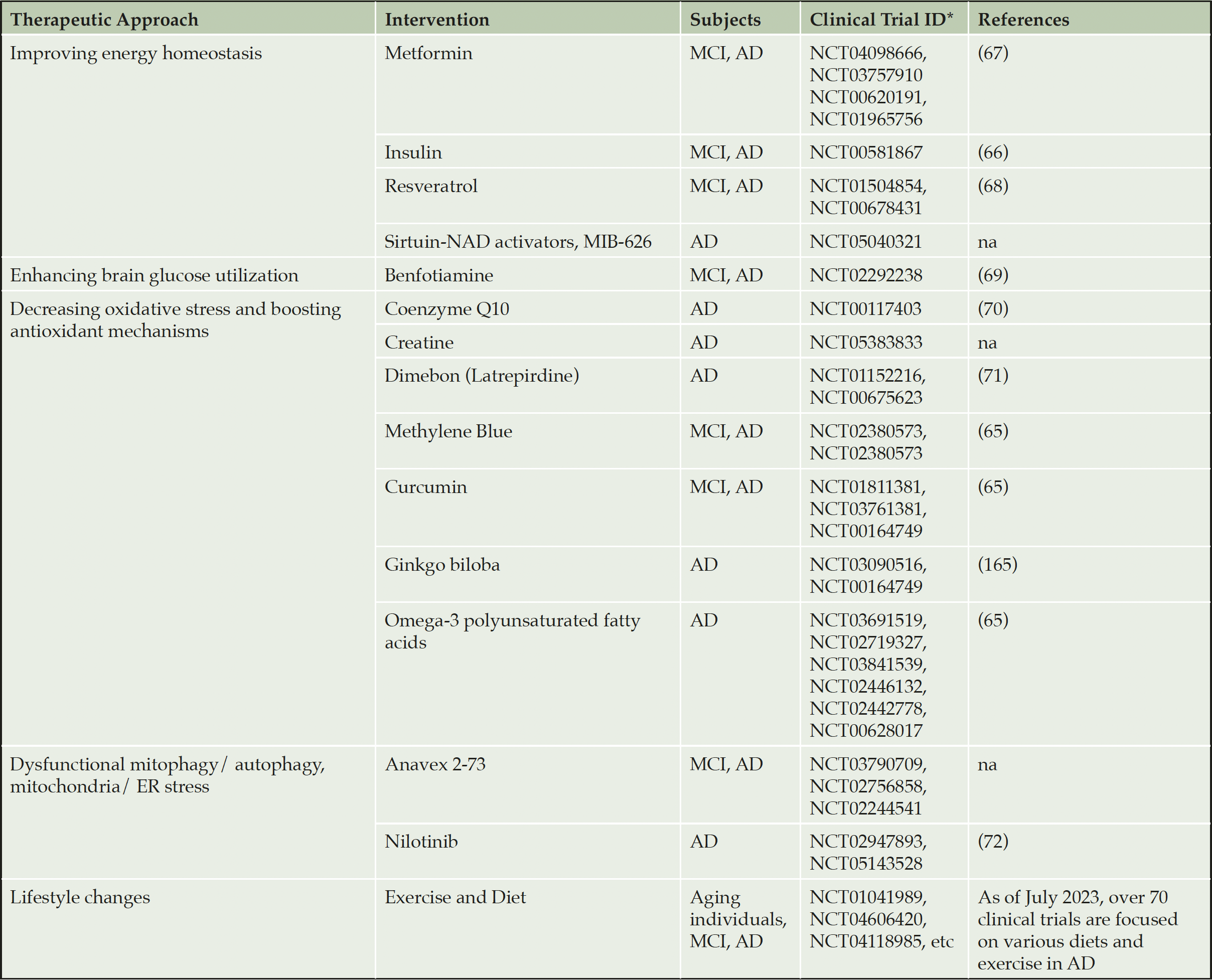
Table 1. Therapeutic strategies that target mitochondrial dysfunction in AD
* Listed are the most recent representative clinical trials. Additional trials could be found on https://clinicaltrials.gov/. na – not available
Mechanisms of adaptive stress response are complex. One of them includes activation of AMPK that has been directly linked to the regulation of cell metabolism, mitochondrial dynamics and function, inflammation, oxidative stress, protein turnover, and Aβ and pTau proteostasis (91-93). Combined analysis performed using multiple types of genome-wide association study (GWAS) data identified a predominant role for metabolism-associated biological processes in the course of AD, including autophagy, insulin and fatty acid metabolism, with a focus on AMPK as a key modulator and therapeutic target (94). Other include hormetic mechanisms such as sublethal ROS production that activates antioxidant response predisposing cells to a better reaction to stress (90). The AMPK-regulated activation of mitochondrial biogenesis and autophagy/mitophagy shifts mitochondria to a younger phenotype improving cellular bioenergetics while simultaneously removing misfolded proteins. Activated AMPK initiates a robust signaling cascade to ensure the restoration of the energy balance. This results in changes in lipid and glucose metabolism, mitochondrial dynamics, mitophagy and biogenesis, protein synthesis, reduction of inflammation and increase in levels of sirtuins, signaling molecules that regulate vast networks of metabolic and non‒metabolic enzymes essential for healthy aging (95-97). While AMPK could activate numerous neuroprotective mechanisms, the approach to develop the direct AMPK activators has been difficult (98). Therefore, the attention has been shifted to indirect AMPK activation via exercise or caloric restriction shown to slow down the progression of NDA (56, 99). Importantly, AMPK activation appears to attenuate major mechanisms involved in AD, including oxidative stress, inflammation, mitochondrial dysfunction, and altered brain energetics (2, 11, 100-102). However, it is important to note that AMPK activation has been also shown to promote Aβ generation and Tau phosphorylation, further emphasizing a fine balance between beneficial and pathological role AMPK may play in NDA (103, 104).
Mild complex I inhibitor CP2
In search for disease-modifying strategies for neurodegenerative diseases, we identified tricyclic pyrone compound (code name CP2) as a mild mitochondrial complex I inhibitor (Fig. 2). To date, efficacy of CP2 treatment has been demonstrated in multiple preclinical mouse models of AD, Huntington’s Disease (HD), and aging (90, 105-109). CP2 penetrates the blood-brain barrier and accumulates in mitochondria where it mildly inhibits the activity of complex I. The specificity of complex I inhibition relative to other OXPHOS complexes was confirmed in mitochondria isolated from a mouse and postmortem human brain tissue (105, 106). The binding of CP2 to ovine complex I has been recently confirmed using Cryo-EM (collaboration with Dr. L. Sazanov, manuscript in preparation).
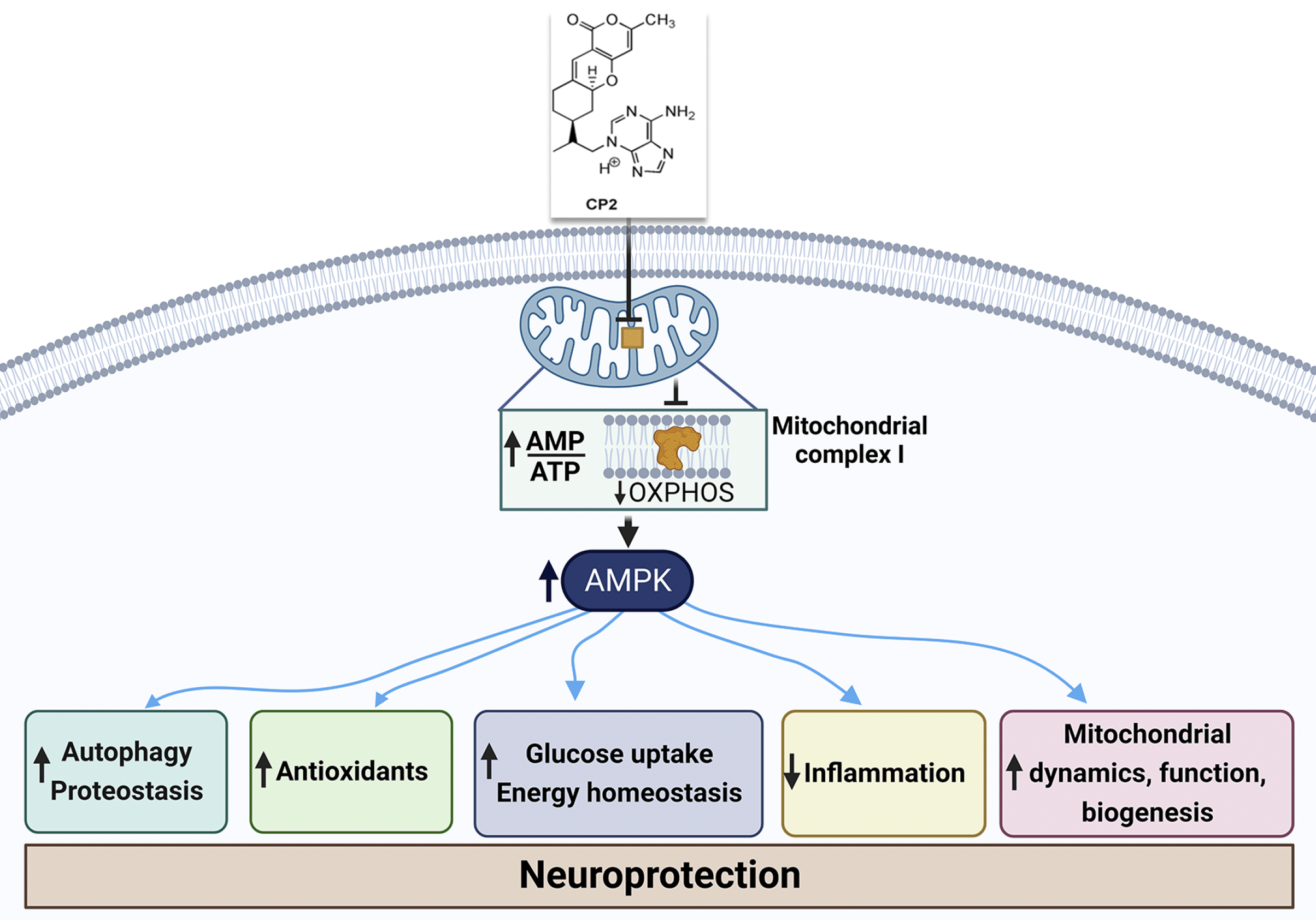
Figure 2. Mild inhibition of mitochondrial complex I activity with small molecule compound CP2 leads to an increase in AMP/ATP ratio that activates AMP-activated protein kinase-dependent mechanisms leading to neuroprotection
AMP, adenosine monophosphate; ATP, adenosine triphosphate. Figure was created with BioRender.com.
CP2 has oral bioavailability of 65% (106). CP2 concentration in the brain tissue of APP/PS1 mice chronically treated from 9 – 23 months of age averaged at ~62 nM, consistent with the earlier bioavailability studies (105, 110-112). Selectivity and specificity of CP2 was evaluated in vitro pharmacological profiling against 44 human targets, including G-protein-coupled receptors, ion channels, enzymes, neurotransmitter transporters, and 250 kinases. At 1 μM and 10 μM, concentrations significantly higher than those found efficacious in mice and in vitro assays, CP2 had minimal off-target activities (106, 109). CP2 (113) was originally identified in the phenotypic screen using MC65 human neuroblastoma cell line engineered to conditionally expresses a 99-residue carboxyl terminal fragment derived from the β-secretase cleavage of human APP, which subsequently is cleaved by γ-secretase to generate Aβ42 (114). Recent comprehensive evaluation demonstrated that intracellular accumulation of Aβ42 in MC65 cells causes multiple pathological mechanisms and induces non-apoptotic cell death via oxytosis/ferroptosis (115). The mechanisms of toxicity included mitochondrial dysfunction associated with altered ATP production, reduced biogenesis, and bioenergetics; increased ROS production; decreased expression of glucose transporters GLUT3, GLUT4 on the plasma membrane; accumulation of Aβ and pTau; inflammation and ER stress (115). CP2 treatment blocked MC65 cell death with EC50 = 150 nM, demonstrating the ability to avert multiple key mechanisms of AD (111, 112). In vitro studies using surface plasmon resonance spectroscopy and atomic force microscopy also demonstrated the ability of CP2 to bind to Aβ and inhibit their aggregation (116). The infusion of 100 µM CP2 into cerebral ventricles of 3-month-old 5x FAD mice resulted in significant reduction of soluble and insoluble Aβ42 (116). In addition to the protection in MC65 cells, CP2 has been shown to inhibit acyl-CoA:cholesterol acyltransferase (ACAT) with EC50 = 1.2 µM and upregulate gene expression of cholesterol transporter ATP-binding cassette subfamily A member 1 (ABCA1) with EC50 = 0.9 mM (117). ACAT is a membrane-bound enzyme that resides at the ER and converts cholesterol to cholesteryl esters (CEs) for storage. Since accumulation of CEs was observed in the brain tissue of AD patients, pharmacological inhibition of ACAT was considered as a therapeutic strategy, and indeed has been shown to reduce levels of Aβ and augment other hallmarks for AD (118). Similarly, the upregulation of ABCA1, which expression and activity are decreased in AD, has been shown beneficial to maintain cholesterol homeostasis (119). However, based on the EC50 values, the effect of CP2 on ACAT or ABCA1 may not be the primary mechanism of protection against Aβ toxicity.
In the past decade, we conducted extensive investigation of CP2 efficacy and molecular mechanisms in cellular and animal models of neurodegenerative diseases and aging. One of the earliest assessments was done in primary embryonic striatal neurons from mouse models of HD that express mutant huntingtin protein (mhtt) (120). We reported that CP2 not only blocked mhtt-induced aggregation in HD neuronal and glial cells, but also alleviated early cellular dysfunctions that preclude aggregate formation in HD neurons, including abnormal cholesterol accumulation and altered clathrin-independent endocytosis (120). Further investigations were conducted in multiple mouse modes of familial AD that express mutant human APP(K670N/M671L), PS1(M146L), double transgenic APP/PS1, and 3xTgAD mice that express mutant human PS1, APPSwe and tauP301L transgenes (105-108). We have shown that chronic administration of CP2 to mice via drinking water ad lib (25 mg/kg/day) was safe over 8 and 14 months of treatment (105, 106, 108). Preliminary toxicology was conducted in breeding wild-type mice treated with 50 mg/kg/day for 30 days (105). Histopathological analysis of multiple tissues from parents and progeny failed to detect abnormalities or the effect on development (105). Administration of CP2 to the breeding APP and PS1 mice and continuous maintenance of progeny on CP2-containing drinking water resulted in a significant delay in the development of AD-like phenotype preserving the performance of CP2-treated APP, PS1 and APP/PS1 mice in multiple behavior tests up to 56 weeks of age (105). Similarly, pre-symptomatic APP/PS1 mice chronically treated with CP2 starting at 2.5 months of age for 6 months maintained the performance in cognitive tests and had reduced Aβ levels in the brain tissue (105). The first indication that CP2 affects mitochondrial respiration was obtained in experiments with a Seahorse Extracellular Flux Analyzer (105). In primary mouse neurons, CP2 was found to accumulate in mitochondria and significantly augment bioenergetic parameters, enhancing the spare respiratory capacity (SRC), the respiratory control ratio, and state apparent. The SRC is an indicator of the mitochondrial ability to produce additional energy under conditions of increased workload or stress, which is essential for a long-term cellular survival and function (121, 122). The SRC can be viewed as a marker of mitochondrial fitness. The fidelity of mitochondrial biogenesis and turnover regulated by a fine balance in the expression of PINK1, Parkin, PGC1α, and the transcription factor A (TFAM), mitochondrial fission/fusion machinery, the efficiency of the ETC and the TCA cycle activity, levels of sirtuins 1 and 3, and activation of AMPK are essential mechanisms for the SRC maintenance. Indeed, we have found that CP2 activated AMPK, increased levels of sirtuin 1 and 3, enhanced mitochondrial biogenesis and turnover in neurons in vitro and in the mouse brain tissue in vivo in the group treated with CP2, compared to the untreated control (105, 106, 108). Collectively, the observed improvement of bioenergetic parameters in CP2-treatd models suggested the tightly coupled ETC and a considerable bioenergetic reserve supporting the improvement of mitochondrial fitness in AD mice.
Furthermore, in primary neurons from PS1 and APP/PS1 mice treated from birth, CP2 treatment restored axonal transport of mitochondria in anterograde (from the cell body) and retrograde (toward the cell body) directions reducing the number of stationary organelles [105]. Inhibition of axonal trafficking is well documented in AD models and in patients (123-126). One of the consequences of the axonal traffic jam is the reduced supply of energy to the sites of synapses and reduced availability of brain derived neurotrophic factor (BDNF), an essential modulator of neuronal health. We have found that in CP2 treated AD mice, levels of BDNF were increased, providing additional support for restoration of mitochondrial dynamics (105, 106). Consistently, improved mitochondrial morphology and increased number of longer organelles, an indication of better bioenergetics, have been found in brain tissue of CP2-treated APP/PS1 mice using electron microscopy (EM) examination (106). Additional effect of CP2 treatment on mitochondria homeostasis included activation of biogenesis and mitophagy, which was also associated with the activation of AMPK (105, 106, 108).
One of the immediate concerns regarding the application of complex I inhibitors is a potential increase in ROS. However, CP2-treated primary cortical neurons exhibited significantly greater resistance to H2O2-induced oxidative damage compared to untreated neurons, which could not be attributed to the CP2 functioning as an antioxidant (105). In APP/PS1 mice chronically treated with CP2 for 14 months starting at 9 months of age, levels of malondialdehyde (MDA), a product of lipid peroxidation, were significantly decreased (106). Experiments in cells demonstrated that the addition of CP2 induced rapid increase of sublethal ROS detected using DCFDA and MitoSOX dyes, which returned to the background levels 60 min later. This ROS did not affect cell viability (unpublished data). To test whether CP2 activates antioxidant response through the mitohormetic ROS signaling, we employed multiple in vitro and in vivo reporter assays (manuscript in preparation). Our data indicate that CP2 activates nuclear factor erythroid 2-related factor 2 (Nrf2) in vitro and in vivo, which further induces the expression of phase II detoxifying enzymes, including heme oxygenase-1 (HO-1), and antioxidant genes through the antioxidant response element (ARE) (manuscript in preparation). This mechanism was ROS-independent and was associated with the activation of the AMPK-p62-KEAP1 axis. Furthermore, CP2 treatment reduced inflammation in AD mouse models. Multiple studies demonstrated that one of the major anti-inflammatory pathways involves activation of the Nrf2 signaling (127). Our data suggest that CP2 treatment reduced inflammation in the brain and blood of APP/PS1 mice, and significantly reduced levels of senescent cells in the adipose tissue of aged non-transgenic (NTG) mice (106).
Consistent with the activation of autophagy, CP2 treatment resulted in reduced levels of Aβ regardless of whether the treatment started at pre- or symptomatic stages of the disease in APP/PS1 mice (105, 106). Significant reduction of human pTau was achieved in male and female 3xTg-AD mice treated with CP2 from 3.5 to 18 months of age (108). This outcome was associated with increased activity of protein phosphatase of type 2A (PP2A), and reduced activity of cyclin-dependent kinase 5 (CDK5) and glycogen synthase kinase 3β (GSK3β). Chronic CP2 treatment restored synaptic activity established in acute brain slices using electrophysiology recording in female 3xTg-AD mice while cognitive function, levels of synaptic proteins, glucose metabolism, and energy homeostasis were improved in male and female 3xTg-AD mice. Similarly, long term potentiation (LTP) was restored in CP2-treated APP/PS1 mice (106). This functional improvement was further confirmed with 3 dimensional reconstructions of dendritic spine morphology using electron micrographs (3D EM). Data supported a presence of mature spines in CP2-treated APP/PS1 mice compared to immature ones observed in the vehicle-treated counterparts. The improvement was also associated with increased levels of synaptic proteins synaptophysin and post-synaptic density protein 95 (PSD95) in the brain tissue of CP2-treated mice (106, 108).
We further examined the effect of CP2 on neuronal loss in the locus coeruleus (LC) of APP/PS1 mice treated after the development of cognitive symptoms and neurodegeneration (106). Like AD patients that exhibit early neurodegeneration in the LC where severity of neuronal loss correlates with the duration of illness, APP/PS1 mice also demonstrate a progressive loss of tyrosine hydroxylase positive (TH+) cortical afferents starting at 6 months of age, followed by the loss of noradrenergic (TH+) neurons in the LC. Consistent with the progressive loss of afferents, there was a reduction in the volume of TH+ neurons in APP/PS1 mice starting at 12 months of age. CP2 treatment started at 9 months of age and delivered for 10 months to APP/PS1 mice completely stopped the progression of neurodegeneration (106). Cortical TH+ axon density, TH+ neuron number in LC, and TH+ neuronal volume in 20-month-old CP2-treated APP/PS1 mice were comparable to those in 12-month-old APP/PS1 mice. These data demonstrate that CP2 specifically protects the neuronal network in APP/PS1 mice, which might be associated with the reduction of Aβ accumulation and toxicity and improved energy homeostasis and mitochondrial function.
Cross-validation of the RNA-seq data generated in APP/PS1 female mice treated with CP2 for 14 months against the human dataset from Accelerating Medicines Partnership in Alzheimer’s Disease (AMP-AD) confirmed translational value of the mouse model and demonstrated that CP2 treatment restored pathways essential for human AD, including inflammation, oxidative stress, and synaptic function (106). Relevant to AD mechanisms, CP2 treatment increased glucose uptake and utilization in the brain of APP/PS1 mice, blocked the ongoing neurodegeneration, and improved mitochondria-ER communication, reducing the ER and UPR stress (106, 107).
Our preclinical studies demonstrated exceptional safety of chronic CP2 administration to mice over extended (up to 20 months) period of time starting in utero (105), at pre-symptomatic (105) or symptomatic stages of the disease (106). To address whether changes in genetic makeup associated with differential gene expression in respect to metabolic pathways and mitochondrial function could preclude from the safe application of this strategy in humans, we conducted a GWAS using a panel of ethnically diversified 196 human lymphoblastoid cell lines (LCLs) that were previously characterized using multi-omics approaches (109, 128). This study provided the first evidence that the application of complex I inhibitor tool compound CP2 was safe, at least in the healthy population. Changes in gene expression in peripheral LCLs overlapped with some of the molecular mechanisms identified in neuronal cells. Genes found to play a potential role in affecting CP2 cytotoxicity were related to drug metabolism, while higher expression of genes involved in antioxidant response appears to increase CP2 tolerance. Except for COX2, no significant associations were found with the expression of the components of mitochondrial OXPHOS machinery, indicating the lack of significant safety concerns with targeting complex I. These results provide strong support for the safe application of complex I inhibitors in a diverse human population at doses shown efficacious in preclinical translational studies (109).
Taken together, our data demonstrate that via mild inhibition of complex I, it is possible to engage multiple neuroprotective mechanisms without targeting each of them individually. This treatment mimics polypharmacy approach that might be necessary to treat complex disorders such as AD. Moreover, data generated in aging mice treated with CP2 suggest that targeting complex I could rejuvenate the transcriptome, promoting healthy aging and delaying the onset of NDA. With support from the Alzheimer Drug Discovery Foundation (ADDF), Harrington Discovery Institute, and the NIH Blueprint program, we have developed novel small molecules mild complex I inhibitors with properties superior to CP2 (three US patents granted). The current stage of drug development is focused on the hit-to-lead optimization and the identification of preclinical drug candidate. While the main focus of the program is to develop safe and efficacious disease-modifying treatment for AD, collaborative data generated in the independent laboratories support feasibility of this treatment for other human conditions such as schizophrenia, rheumatoid arthritis, polycystic kidney disease, and osteoarthritis. Collectively, these data indicate that harnessing mitochondrial signaling could be beneficial for multiple human diseases granting further exploration of this therapeutic approach.
Funding: This research was supported by the grants from the National Institutes of Health grant numbers RF1AG55549, RO1AG062135, AG59093), Harrington Discovery Institute and Alzheimer Drug Discovery Foundation (all to ET). Dr. Thi Kim Oanh Nguyen is supported by the Alzheimer’s Association Research Fellowship grant # 23AARF-1027342. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NIH and other funding organizations. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgement: We thank Ms. S. Gochnauer for help with the manuscript formatting and Dr. H. Lee for editing the manuscript.
Conflict of interest: Dr. Trushina and Mayo Clinic hold three US patents on novel small molecule mitochondria targeted compounds. The authors declare no conflict of interest.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol 2013, 12, 357-367, doi:10.1016/S1474-4422(13)70044-9.
2. Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov 2020, 19, 609-633, doi:10.1038/s41573-020-0072-x.
3. Tzioras, M.; McGeachan, R.I.; Durrant, C.S.; Spires-Jones, T.L. Synaptic degeneration in Alzheimer disease. Nat Rev Neurol 2023, 19, 19-38, doi:10.1038/s41582-022-00749-z.
4. Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient invasions: from endosymbionts to organelles. Science 2004, 304, 253-257, doi:10.1126/science.1094884.
5. Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 2020, 21, 204-224, doi:10.1038/s41580-020-0210-7.
6. Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194-1217, doi:10.1016/j.cell.2013.05.039.
7. Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335-343, doi:10.1038/nature12985.
8. Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 2011, 14, 724-738, doi:10.1016/j.cmet.2011.08.016.
9. Vacanti, N.M.; Divakaruni, A.S.; Green, C.R.; Parker, S.J.; Henry, R.R.; Ciaraldi, T.P.; Murphy, A.N.; Metallo, C.M. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol Cell 2014, 56, 425-435, doi:10.1016/j.molcel.2014.09.024.
10. Huttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J Bioenerg Biomembr 2008, 40, 445-456, doi:10.1007/s10863-008-9169-3.
11. Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J Alzheimers Dis 2017, 57, 1105-1121, doi:10.3233/JAD-161088.
12. Brand, M.D.; Chien, L.F.; Ainscow, E.K.; Rolfe, D.F.; Porter, R.K. The causes and functions of mitochondrial proton leak. Biochim Biophys Acta 1994, 1187, 132-139.
13. Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in Mitochondrial Dynamics and Metabolomic Signatures of Evolving Energetic Stress in Mouse Models of Familial Alzheimer’s Disease. PLoS One 2012, 7, doi:ARTN e32737, DOI 10.1371/journal.pone.0032737.
14. Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener 2020, 15, 30, doi:10.1186/s13024-020-00376-6.
15. Michel, S.; Wanet, A.; De Pauw, A.; Rommelaere, G.; Arnould, T.; Renard, P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J Cell Physiol 2012, 227, 2297-2310, doi:10.1002/jcp.23021.
16. Zhang, L.; Wu, J.; Zhu, Z.; He, Y.; Fang, R. Mitochondrion: A bridge linking aging and degenerative diseases. Life Sci 2023, 322, 121666, doi:10.1016/j.lfs.2023.121666.
17. Grimm, M.O.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front Mol Neurosci 2017, 10, 63, doi:10.3389/fnmol.2017.00063.
18. Chandel, N.S. Evolution of Mitochondria as Signaling Organelles. Cell Metab 2015, 22, 204-206, doi:10.1016/j.cmet.2015.05.013.
19. Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 2009, 20, 98-105, doi:10.1097/MOL.0b013e328328d0a4.
20. Ye, Q.; Huang, W.; Li, D.; Si, E.; Wang, J.; Wang, Y.; Chen, C.; Chen, X. Overexpression of PGC-1alpha Influences Mitochondrial Signal Transduction of Dopaminergic Neurons. Mol Neurobiol 2016, 53, 3756-3770, doi:10.1007/s12035-015-9299-7.
21. Mota, B.C.; Sastre, M. The Role of GWAS lpha in Alzheimer’s Disease and Therapeutic Interventions. Int J Mol Sci 2021, 22, doi:10.3390/ijms22115769.
22. Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011, 13, 132-141, doi:10.1038/ncb2152.
23. Aman, Y.; Frank, J.; Lautrup, S.H.; Matysek, A.; Niu, Z.; Yang, G.; Shi, L.; Bergersen, L.H.; Storm-Mathisen, J.; Rasmussen, L.J.; et al. The NAD(+)-mitophagy axis in healthy longevity and in artificial intelligence-based clinical applications. Mech Ageing Dev 2020, 185, 111194, doi:10.1016/j.mad.2019.111194.
24. Kataura, T.; Sedlackova, L.; Otten, E.G.; Kumari, R.; Shapira, D.; Scialo, F.; Stefanatos, R.; Ishikawa, K.I.; Kelly, G.; Seranova, E.; et al. Autophagy promotes cell survival by maintaining NAD levels. Dev Cell 2022, 57, 2584-2598 e2511, doi:10.1016/j.devcel.2022.10.008.
25. Quinn, P.M.J.; Moreira, P.I.; Ambrosio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol Commun 2020, 8, 189, doi:10.1186/s40478-020-01062-w.
26. Killackey, S.A.; Philpott, D.J.; Girardin, S.E. Mitophagy pathways in health and disease. J Cell Biol 2020, 219, doi:10.1083/jcb.202004029.
27. Xie, C.; Zhuang, X.X.; Niu, Z.; Ai, R.; Lautrup, S.; Zheng, S.; Jiang, Y.; Han, R.; Gupta, T.S.; Cao, S.; et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat Biomed Eng 2022, 6, 76-93, doi:10.1038/s41551-021-00819-5.
28. Rose, G.; Santoro, A.; Salvioli, S. Mitochondria and mitochondria-induced signalling molecules as longevity determinants. Mech Ageing Dev 2017, 165, 115-128, doi:10.1016/j.mad.2016.12.002.
29. Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt). Cell 2016, 165, 1197-1208, doi:10.1016/j.cell.2016.04.011.
30. Lin, M.M.; Liu, N.; Qin, Z.H.; Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol Sin 2022, 43, 2439-2447, doi:10.1038/s41401-022-00879-6.
31. Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab 2015, 21, 443-454, doi:10.1016/j.cmet.2015.02.009.
32. Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531-1536, doi:10.1126/science.aav4011.
33. West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553-557, doi:10.1038/nature14156.
34. Benayoun, B.A.; Lee, C. MOTS-c: A Mitochondrial-Encoded Regulator of the Nucleus. Bioessays 2019, 41, e1900046, doi:10.1002/bies.201900046.
35. Peng, T.; Xie, Y.; Sheng, H.; Wang, C.; Lian, Y.; Xie, N. Mitochondrial-derived vesicles: Gatekeepers of mitochondrial response to oxidative stress. Free Radic Biol Med 2022, 188, 185-193, doi:10.1016/j.freeradbiomed.2022.06.233.
36. Lin, M.Y.; Cheng, X.T.; Tammineni, P.; Xie, Y.; Zhou, B.; Cai, Q.; Sheng, Z.H. Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 2017, 94, 595-610 e596, doi:10.1016/j.neuron.2017.04.004.
37. Deus, C.M.; Tavares, H.; Beatriz, M.; Mota, S.; Lopes, C. Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders. Cells 2022, 11, doi:10.3390/cells11152364.
38. Crawford, K.; Leonenko, G.; Baker, E.; Grozeva, D.; Lan-Leung, B.; Holmans, P.; Williams, J.; O’Donovan, M.C.; Escott-Price, V.; Ivanov, D.K. Golgi apparatus, endoplasmic reticulum and mitochondrial function implicated in Alzheimer’s disease through polygenic risk and RNA sequencing. Mol Psychiatry 2023, 28, 1327-1336, doi:10.1038/s41380-022-01926-8.
39. Barazzuol, L.; Giamogante, F.; Cali, T. Mitochondria Associated Membranes (MAMs): Architecture and physiopathological role. Cell Calcium 2021, 94, 102343, doi:10.1016/j.ceca.2020.102343.
40. Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res Bull 2017, 133, 71-79, doi:10.1016/j.brainresbull.2016.08.009.
41. Gabrielli, A.P.; Weidling, I.; Ranjan, A.; Wang, X.; Novikova, L.; Chowdhury, S.R.; Menta, B.; Berkowicz, A.; Wilkins, H.M.; Peterson, K.R.; et al. Mitochondria Profoundly Influence Apolipoprotein E Biology. J Alzheimers Dis 2023, doi:10.3233/JAD-221177.
42. Samluk, L.; Ostapczuk, P.; Dziembowska, M. Long-term mitochondrial stress induces early steps of Tau aggregation by increasing reactive oxygen species levels and affecting cellular proteostasis. Mol Biol Cell 2022, 33, ar67, doi:10.1091/mbc.E21-11-0553.
43. Bennett, J.P., Jr.; Keeney, P.M. Alzheimer’s and Parkinson’s brain tissues have reduced expression of genes for mtDNA OXPHOS Proteins, mitobiogenesis regulator PGC-1alpha protein and mtRNA stabilizing protein LRPPRC (LRP130). Mitochondrion 2020, 53, 154-157, doi:10.1016/j.mito.2020.05.012.
44. Trumpff, C.; Owusu-Ansah, E.; Klein, H.U.; Lee, A.J.; Petyuk, V.; Wingo, T.S.; Wingo, A.P.; Thambisetty, M.; Ferrucci, L.; Seyfried, N.T.; et al. Mitochondrial respiratory chain protein co-regulation in the human brain. Heliyon 2022, 8, e09353, doi:10.1016/j.heliyon.2022.e09353.
45. Wingo, A.P.; Dammer, E.B.; Breen, M.S.; Logsdon, B.A.; Duong, D.M.; Troncosco, J.C.; Thambisetty, M.; Beach, T.G.; Serrano, G.E.; Reiman, E.M.; et al. Large-scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat Commun 2019, 10, 1619, doi:10.1038/s41467-019-09613-z.
46. Klein, H.U.; Trumpff, C.; Yang, H.S.; Lee, A.J.; Picard, M.; Bennett, D.A.; De Jager, P.L. Characterization of mitochondrial DNA quantity and quality in the human aged and Alzheimer’s disease brain. Mol Neurodegener 2021, 16, 75, doi:10.1186/s13024-021-00495-8.
47. Salminen, A.; Haapasalo, A.; Kauppinen, A.; Kaarniranta, K.; Soininen, H.; Hiltunen, M. Impaired mitochondrial energy metabolism in Alzheimer’s disease: Impact on pathogenesis via disturbed epigenetic regulation of chromatin landscape. Prog Neurobiol 2015, 131, 1-20, doi:10.1016/j.pneurobio.2015.05.001.
48. Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 2008, 31, 454-463.
49. Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 2018, 4, 575-590, doi:10.1016/j.trci.2018.06.014.
50. Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J Alzheimers Dis 2018, 62, 1403-1416, doi:10.3233/JAD-170585.
51. Trushina, E.; Dutta, T.; Persson, X.M.; Mielke, M.M.; Petersen, R.C. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS One 2013, 8, e63644, doi:10.1371/journal.pone.0063644.
52. Leuner, K.; Schulz, K.; Schutt, T.; Pantel, J.; Prvulovic, D.; Rhein, V.; Savaskan, E.; Czech, C.; Eckert, A.; Muller, W.E. Peripheral mitochondrial dysfunction in Alzheimer’s disease: focus on lymphocytes. Molecular neurobiology 2012, 46, 194-204, doi:10.1007/s12035-012-8300-y.
53. Perez, M.J.; Ponce, D.P.; Osorio-Fuentealba, C.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial Bioenergetics Is Altered in Fibroblasts from Patients with Sporadic Alzheimer’s Disease. Front Neurosci 2017, 11, 553, doi:10.3389/fnins.2017.00553.
54. Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov 2018, 17, 865-886, doi:10.1038/nrd.2018.174.
55. Mi, Y.; Qi, G.; Brinton, R.D.; Yin, F. Mitochondria-Targeted Therapeutics for Alzheimer’s Disease: The Good, the Bad, the Potential. Antioxid Redox Signal 2021, 34, 611-630, doi:10.1089/ars.2020.8070.
56. Flannery, P.J.; Trushina, E. Mitochondrial Dysfunction in Alzheimer’s Disease and Progress in Mitochondria-Targeted Therapeutics. Current Behavioral Neuroscience Reports 2019, 6, 88–102, doi:https://doi.org/10.1007/s40473-019-00179-0.
57. Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front Pharmacol 2015, 6, 206, doi:10.3389/fphar.2015.00206.
58. Xu, J.; Du, W.; Zhao, Y.; Lim, K.; Lu, L.; Zhang, C.; Li, L. Mitochondria targeting drugs for neurodegenerative diseases-Design, mechanism and application. Acta Pharm Sin B 2022, 12, 2778-2789, doi:10.1016/j.apsb.2022.03.001.
59. Plascencia-Villa, G.; Perry, G. Exploring Molecular Targets for Mitochondrial Therapies in Neurodegenerative Diseases. Int J Mol Sci 2023, 24, doi:10.3390/ijms241512486.
60. Kellar, D.; Register, T.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal insulin modulates cerebrospinal fluid markers of neuroinflammation in mild cognitive impairment and Alzheimer’s disease: a randomized trial. Sci Rep 2022, 12, 1346, doi:10.1038/s41598-022-05165-3.
61. Luchsinger, J.A.; Perez, T.; Chang, H.; Mehta, P.; Steffener, J.; Pradabhan, G.; Ichise, M.; Manly, J.; Devanand, D.P.; Bagiella, E. Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J Alzheimers Dis 2016, 51, 501-514, doi:10.3233/JAD-150493.
62. Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J Neuroinflammation 2017, 14, 1, doi:10.1186/s12974-016-0779-0.
63. Gibson, G.E.; Luchsinger, J.A.; Cirio, R.; Chen, H.; Franchino-Elder, J.; Hirsch, J.A.; Bettendorff, L.; Chen, Z.; Flowers, S.A.; Gerber, L.M.; et al. Benfotiamine and Cognitive Decline in Alzheimer’s Disease: Results of a Randomized Placebo-Controlled Phase IIa Clinical Trial. J Alzheimers Dis 2020, 78, 989-1010, doi:10.3233/JAD-200896.
64. Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol 2012, 69, 836-841, doi:10.1001/archneurol.2012.85.
65. Cano-Cuenca, N.; Solis-Garcia del Pozo, J.E.; Jordan, J. Evidence for the efficacy of latrepirdine (Dimebon) treatment for improvement of cognitive function: a meta-analysis. J Alzheimers Dis 2014, 38, 155-164, doi:10.3233/JAD-130872.
66. Turner, R.S.; Hebron, M.L.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, J.N.; Anjum, M.; Pagan, F.; Torres-Yaghi, Y.; Shi, W.; et al. Nilotinib Effects on Safety, Tolerability, and Biomarkers in Alzheimer’s Disease. Ann Neurol 2020, 88, 183-194, doi:10.1002/ana.25775.
67. Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337-342, doi:10.1038/nature05354.
68. Greco, S.J.; Hamzelou, A.; Johnston, J.M.; Smith, M.A.; Ashford, J.W.; Tezapsidis, N. Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and beta-amyloid in neurons. Biochemical and biophysical research communications 2011, 414, 170-174, doi:10.1016/j.bbrc.2011.09.050.
69. Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Current biology : CB 2007, 17, 1646-1656, doi:10.1016/j.cub.2007.08.047.
70. Mair, W.; Morantte, I.; Rodrigues, A.P.; Manning, G.; Montminy, M.; Shaw, R.J.; Dillin, A. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 2011, 470, 404-408, doi:10.1038/nature09706.
71. Pani, G. Neuroprotective effects of dietary restriction: Evidence and mechanisms. Semin Cell Dev Biol 2015, 40, 106-114, doi:10.1016/j.semcdb.2015.03.004.
72. Porquet, D.; Casadesus, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegri, C.; Sanfeliu, C.; Camins, A.; Pallas, M.; et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age (Dordr) 2013, 35, 1851-1865, doi:10.1007/s11357-012-9489-4.
73. Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288-341, doi:10.2203/dose-response.13-035.Ristow.
74. Baumgart, M.; Priebe, S.; Groth, M.; Hartmann, N.; Menzel, U.; Pandolfini, L.; Koch, P.; Felder, M.; Ristow, M.; Englert, C.; et al. Longitudinal RNA-Seq Analysis of Vertebrate Aging Identifies Mitochondrial Complex I as a Small-Molecule-Sensitive Modifier of Lifespan. Cell Syst 2016, 2, 122-132, doi:10.1016/j.cels.2016.01.014.
75. Feng, J.; Bussiere, F.; Hekimi, S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Developmental cell 2001, 1, 633-644.
76. Raule, N.; Sevini, F.; Li, S.; Barbieri, A.; Tallaro, F.; Lomartire, L.; Vianello, D.; Montesanto, A.; Moilanen, J.S.; Bezrukov, V.; et al. The co-occurrence of mtDNA mutations on different oxidative phosphorylation subunits, not detected by haplogroup analysis, affects human longevity and is population specific. Aging Cell 2014, 13, 401-407, doi:10.1111/acel.12186.
77. Yang, W.; Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010, 9, 433-447, doi:10.1111/j.1474-9726.2010.00571.x.
78. Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS biology 2010, 8, e1000556, doi:10.1371/journal.pbio.1000556.
79. Chen, Y.; Zhao, S.; Fan, Z.; Li, Z.; Zhu, Y.; Shen, T.; Li, K.; Yan, Y.; Tian, J.; Liu, Z.; et al. Metformin attenuates plaque-associated tau pathology and reduces amyloid-beta burden in APP/PS1 mice. Alzheimers Res Ther 2021, 13, 40, doi:10.1186/s13195-020-00761-9.
80. Copeland, J.M.; Cho, J.; Lo, T., Jr.; Hur, J.H.; Bahadorani, S.; Arabyan, T.; Rabie, J.; Soh, J.; Walker, D.W. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Current biology : CB 2009, 19, 1591-1598, doi:10.1016/j.cub.2009.08.016.
81. Owusu-Ansah, E.; Song, W.; Perrimon, N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 2013, 155, 699-712, doi:10.1016/j.cell.2013.09.021.
82. Bridges, H.R.; Blaza, J.N.; Yin, Z.; Chung, I.; Pollak, M.N.; Hirst, J. Structural basis of mammalian respiratory complex I inhibition by medicinal biguanides. Science 2023, 379, 351-357, doi:10.1126/science.ade3332.
83. Hu, D.; Xie, F.; Xiao, Y.; Lu, C.; Zhong, J.; Huang, D.; Chen, J.; Wei, J.; Jiang, Y.; Zhong, T. Metformin: A Potential Candidate for Targeting Aging Mechanisms. Aging Dis 2021, 12, 480-493, doi:10.14336/AD.2020.0702.
84. Sluggett, J.K.; Koponen, M.; Bell, J.S.; Taipale, H.; Tanskanen, A.; Tiihonen, J.; Uusitupa, M.; Tolppanen, A.M.; Hartikainen, S. Metformin and Risk of Alzheimer’s Disease Among Community-Dwelling People With Diabetes: A National Case-Control Study. J Clin Endocrinol Metab 2020, 105, doi:10.1210/clinem/dgz234.
85. Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab 2020, 32, 15-30, doi:10.1016/j.cmet.2020.04.001.
86. Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep 2019, 29, 1511-1523 e1515, doi:10.1016/j.celrep.2019.09.070.
87. Cameron, A.R.; Logie, L.; Patel, K.; Erhardt, S.; Bacon, S.; Middleton, P.; Harthill, J.; Forteath, C.; Coats, J.T.; Kerr, C.; et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol 2018, 14, 187-197, doi:10.1016/j.redox.2017.08.018.
88. Poor, S.R.; Ettcheto, M.; Cano, A.; Sanchez-Lopez, E.; Manzine, P.R.; Olloquequi, J.; Camins, A.; Javan, M. Metformin a Potential Pharmacological Strategy in Late Onset Alzheimer’s Disease Treatment. Pharmaceuticals (Basel) 2021, 14, doi:10.3390/ph14090890.
89. Khezri, M.R.; Yousefi, K.; Mahboubi, N.; Hodaei, D.; Ghasemnejad-Berenji, M. Metformin in Alzheimer’s disease: An overview of potential mechanisms, preclinical and clinical findings. Biochem Pharmacol 2022, 197, 114945, doi:10.1016/j.bcp.2022.114945.
90. Trushina, E.; Trushin, S.; Hasan, M.F. Mitochondrial complex I as a therapeutic target for Alzheimer’s disease. Acta Pharm Sin B 2022, 12, 483-495, doi:10.1016/j.apsb.2021.11.003.
91. Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev 2012, 11, 230-241, doi:10.1016/j.arr.2011.12.005.
92. Park, S.; Artan, M.; Han, S.H.; Park, H.H.; Jung, Y.; Hwang, A.B.; Shin, W.S.; Kim, K.T.; Lee, S.V. VRK-1 extends life span by activation of AMPK via phosphorylation. Sci Adv 2020, 6, doi:10.1126/sciadv.aaw7824.
93. Zhang, Y.; Lanjuin, A.; Chowdhury, S.R.; Mistry, M.; Silva-Garcia, C.G.; Weir, H.J.; Lee, C.L.; Escoubas, C.C.; Tabakovic, E.; Mair, W.B. Neuronal TORC1 modulates longevity via AMPK and cell nonautonomous regulation of mitochondrial dynamics in C. elegans. Elife 2019, 8, doi:10.7554/eLife.49158.
94. Caberlotto, L.; Lauria, M.; Nguyen, T.P.; Scotti, M. The central role of AMP-kinase and energy homeostasis impairment in Alzheimer’s disease: a multifactor network analysis. PLoS One 2013, 8, e78919, doi:10.1371/journal.pone.0078919.
95. Peixoto, C.A.; Oliveira, W.H.; Araujo, S.; Nunes, A.K.S. AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration. Exp Neurol 2017, 298, 31-41, doi:10.1016/j.expneurol.2017.08.013.
96. Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab 2010, 298, E751-760, doi:10.1152/ajpendo.00745.2009.
97. van de Ven, R.A.H.; Santos, D.; Haigis, M.C. Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol Med 2017, 23, 320-331, doi:10.1016/j.molmed.2017.02.005.
98. Steinberg, G.R.; Carling, D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov 2019, doi:10.1038/s41573-019-0019-2.
99. Broskey, N.T.; Marlatt, K.L.; Most, J.; Erickson, M.L.; Irving, B.A.; Redman, L.M. The Panacea of Human Aging: Calorie Restriction Versus Exercise. Exerc Sport Sci Rev 2019, 47, 169-175, doi:10.1249/JES.0000000000000193.
100. Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov 2021, 20, 689-709, doi:10.1038/s41573-021-00233-1.
101. Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp Neurol 2020, 330, 113321, doi:10.1016/j.expneurol.2020.113321.
102. Mosconi, L.; Pupi, A.; De Leon, M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Annals of the New York Academy of Sciences 2008, 1147, 180-195, doi:10.1196/annals.1427.007.
103. Cai, Z.; Yan, L.J.; Li, K.; Quazi, S.H.; Zhao, B. Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromolecular Med 2012, 14, 1-14, doi:10.1007/s12017-012-8173-2.
104. Assefa, B.T.; Tafere, G.G.; Wondafrash, D.Z.; Gidey, M.T. The Bewildering Effect of AMPK Activators in Alzheimer’s Disease: Review of the Current Evidence. Biomed Res Int 2020, 2020, 9895121, doi:10.1155/2020/9895121.
105. Zhang, L.; Zhang, S.; Maezawa, I.; Trushin, S.; Minhas, P.; Pinto, M.; Jin, L.W.; Prasain, K.; Nguyen, T.D.; Yamazaki, Y.; et al. Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s Disease. EBioMedicine 2015, 2, 294-305, doi:10.1016/j.ebiom.2015.03.009.
106. Stojakovic, A.; Trushin, S.; Sheu, A.; Khalili, L.; Chang, S.Y.; Li, X.; Christensen, T.; Salisbury, J.L.; Geroux, R.E.; Gateno, B.; et al. Partial inhibition of mitochondrial complex I ameliorates Alzheimer’s disease pathology and cognition in APP/PS1 female mice. Commun Biol 2021, 4, 61, doi:10.1038/s42003-020-01584-y.
107. Panes, J.; Nguyen, t.K.O.; Gao, H.; Christensen, T.A.; Stojakovic, A.; Trushin, S.; Salisbury, J.L.; Fuentealba, J.; Trushina, E. Partial Inhibition of Complex I Restores Mitochondrial Morphology and Mitochondria-ER Communication in Hippocampus of APP/PS1 Mice. Cells 2023, 12, doi:https://doi.org/10.3390/cells12081111.
108. Stojakovic, A.; Chang, S.Y.; Nesbitt, J.; Pichurin, N.P.; Ostroot, M.A.; Aikawa, T.; Kanekiyo, T.; Trushina, E. Partial Inhibition of Mitochondrial Complex I Reduces Tau Pathology and Improves Energy Homeostasis and Synaptic Function in 3xTg-AD Mice. J Alzheimers Dis 2021, 79, 335-353, doi:10.3233/JAD-201015.
109. Gao, H.; Tripathi, U.; Trushin, S.; Okromelidze, L.; Pichurin, N.P.; Wei, L.; Zhuang, Y.; Wang, L.; Trushina, E. A Genome-Wide Association Study in Human Lymphoblastoid Cells Supports Safety of Mitochondrial Complex I Inhibitor. Mitochondrion 2021, In Press, doi:10.1016/j.mito.2021.02.005.
110. Zhang, L.; Zhang, S.; Maezawa, I.; Trushin, S.; Minhas, P.; Pinto, M.; Jin, L.W.; Prasain, K.; Nguyen, T.D.T.; Yamazaki, Y.; et al. Corrigendum to “Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease” [EBioMedicine 2 (2015) 294-305]. EBioMedicine 2019, 42, 532, doi:10.1016/j.ebiom.2019.03.062.
111. Jin, L.W.; Hua, D.H.; Shie, F.S.; Maezawa, I.; Sopher, B.; Martin, G.M. Novel tricyclic pyrone compounds prevent intracellular APP C99-induced cell death. J Mol Neurosci 2002, 19, 57-61, doi:JMN:19:1-2:57 [pii] 10.1007/s12031-002-0011-9.
112. Maezawa, I.; Hong, H.S.; Wu, H.C.; Battina, S.K.; Rana, S.; Iwamoto, T.; Radke, G.A.; Pettersson, E.; Martin, G.M.; Hua, D.H.; et al. A novel tricyclic pyrone compound ameliorates cell death associated with intracellular amyloid-beta oligomeric complexes. J Neurochem 2006, 98, 57-67, doi:10.1111/j.1471-4159.2006.03862.x.
113. Hua, D.H.; Huang, X.; Tamura, M.; Chen, Y.; Woltkamp, M.; Jin, L.; Perchellet, E.M.; Perchellet, J.; Chiang, P.K.; Namatame, I.; et al. Syntheses and bioactivities of tricyclic pyrones. Tetrahedron 2003, 4795–4803.
114. Sopher, B.L.; Fukuchi, K.; Smith, A.C.; Leppig, K.A.; Furlong, C.E.; Martin, G.M. Cytotoxicity mediated by conditional expression of a carboxyl-terminal derivative of the beta-amyloid precursor protein. Brain Res Mol Brain Res 1994, 26, 207-217, doi:10.1016/0169-328x(94)90092-2.
115. Huang, L.; McClatchy, D.B.; Maher, P.; Liang, Z.; Diedrich, J.K.; Soriano-Castell, D.; Goldberg, J.; Shokhirev, M.; Yates, J.R., 3rd; Schubert, D.; et al. Intracellular amyloid toxicity induces oxytosis/ferroptosis regulated cell death. Cell Death Dis 2020, 11, 828, doi:10.1038/s41419-020-03020-9.
116. Hong, H.S.; Rana, S.; Barrigan, L.; Shi, A.; Zhang, Y.; Zhou, F.; Jin, L.W.; Hua, D.H. Inhibition of Alzheimer’s amyloid toxicity with a tricyclic pyrone molecule in vitro and in vivo. J Neurochem 2009, 108, 1097-1108, doi:10.1111/j.1471-4159.2008.05866.x.
117. Pokhrel, L.; Maezawa, I.; Nguyen, T.D.; Chang, K.O.; Jin, L.W.; Hua, D.H. Inhibition of Acyl-CoA: cholesterol acyltransferase (ACAT), overexpression of cholesterol transporter gene, and protection of amyloid beta (Abeta) oligomers-induced neuronal cell death by tricyclic pyrone molecules. J Med Chem 2012, 55, 8969-8973, doi:10.1021/jm3012189.
118. Chang, T.Y.; Chang, C.C.Y.; Harned, T.C.; De La Torre, A.L.; Lee, J.; Huynh, T.N.; Gow, J.G. Blocking cholesterol storage to treat Alzheimer’s disease. Explor Neuroprotective Ther 2021, 1, 173-184, doi:10.37349/ent.2021.00014.
119. Lewandowski, C.T.; Laham, M.S.; Thatcher, G.R.J. Remembering your A, B, C’s: Alzheimer’s disease and ABCA1. Acta Pharm Sin B 2022, 12, 995-1018, doi:10.1016/j.apsb.2022.01.011.
120. Trushina, E.; Rana, S.; McMurray, C.T.; Hua, D.H. Tricyclic pyrone compounds prevent aggregation and reverse cellular phenotypes caused by expression of mutant huntingtin protein in striatal neurons. BMC Neurosci 2009, 10, 73, doi:1471-2202-10-73 [pii], 10.1186/1471-2202-10-73.
121. Choi, S.W.; Gerencser, A.A.; Nicholls, D.G. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. J Neurochem 2009, 109, 1179-1191, doi:10.1111/j.1471-4159.2009.06055.x.
122. Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J 2020, 34, 13106-13124, doi:10.1096/fj.202000767R.
123. Zhang, L.; Trushin, S.; Christensen, T.A.; Tripathi, U.; Hong, C.; Geroux, R.E.; Howell, K.G.; Poduslo, J.F.; Trushina, E. Differential effect of amyloid beta peptides on mitochondrial axonal trafficking depends on their state of aggregation and binding to the plasma membrane. Neurobiol Dis 2018, 114, 1-16, doi:10.1016/j.nbd.2018.02.003.
124. Correia, S.C.; Perry, G.; Moreira, P.I. Mitochondrial traffic jams in Alzheimer’s disease – pinpointing the roadblocks. Biochim Biophys Acta 2016, 1862, 1909-1917, doi:10.1016/j.bbadis.2016.07.010.
125. Ramser, E.M.; Gan, K.J.; Decker, H.; Fan, E.Y.; Suzuki, M.M.; Ferreira, S.T.; Silverman, M.A. Amyloid-beta oligomers induce tau-independent disruption of BDNF axonal transport via calcineurin activation in cultured hippocampal neurons. Mol Biol Cell 2013, 24, 2494-2505, doi:10.1091/mbc.E12-12-0858.
126. Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci 2013, 14, 161-176, doi:10.1038/nrn3380.
127. Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim Biophys Acta Mol Basis Dis 2017, 1863, 585-597, doi:10.1016/j.bbadis.2016.11.005.
128. Niu, N.; Wang, L. In vitro human cell line models to predict clinical response to anticancer drugs. Pharmacogenomics 2015, 16, 273-285, doi:10.2217/pgs.14.170.
© The Authors 2023