
E. Vijverberg1, W. de Haan1, E. Scheijbeler1, M.E. Hamby2, S. Catalano3, P. Scheltens1, M. Grundman4,5, A.O. Caggiano2
1. Alzheimer Center Amsterdam, Neurology, Vrije Universiteit Amsterdam, Amsterdam UMC location VUmc, Amsterdam, The Netherlands; 2. Cognition Therapeutics, Inc., Purchase, NY, USA; 3. Capsida Biotherapeutics, Thousand Oaks, CA, employee of Cognition Therapeutics, Inc. at the time of this study, 4. Global R&D Partners, LLC, San Diego, California, USA; 5. Department of Neurosciences, University of California, San Diego, USA
Corresponding Author: Anthony O. Caggiano, MD, PhD, Cognition Therapeutics, Inc., 2500 Westchester Avenue, Purchase, NY 10577, acaggiano@cogrx.com
J Prev Alz Dis 2024;6(11):1809-1817
Published online August 7, 2024, http://dx.doi.org/10.14283/jpad.2024.154
Abstract
BACKGROUND: CT1812 is a first-in-class, sigma-2 receptor ligand, that prevents and displaces binding of amyloid beta (Aβ) oligomers. Normalization of quantitative electroencephalography (qEEG) markers suggests that CT1812 protects synapses from Aβ oligomer toxicity.
OBJECTIVES: Evaluate CT1812 impact on synaptic function using qEEG measurements.
DESIGN: Phase 2, randomized, double-blind, placebo-controlled, 4-week crossover study.
SETTING: VU University Medical Center and Brain Research Center Amsterdam, The Netherlands.
PARTICIPANTS: Adults with mild or moderate Alzheimer’s disease (AD).
INTERVENTION: A daily 300 mg dose of CT1812 or placebo for 4 weeks.
MEASUREMENTS: A resting-state, eyes closed qEEG assessment occurred on Day 1 and on Day 29 of Treatment Periods 1 and 2, and at follow-up. The primary endpoint was global relative theta power (4-8 Hz), along with secondary EEG measures including global alpha corrected Amplitude Envelope Correlation (AEC-c). Cognitive and functional assessments, fluid biomarkers, and safety and tolerability were assessed.
RESULTS: 16 patients were randomized, and 15 completed. A non-significant (p=0.123) but consistent reduction occurred in global relative theta power and in relative theta power in frontal, temporal, parietal, occipital and central (p<0.006) brain regions with CT1812. A nominally significant (p=0.034) improvement was observed in global alpha AEC-c. Adverse events occurred in 11 patients with CT1812 and 6 with placebo – most commonly nausea, diarrhea, and procedural headache. No severe or serious AEs, deaths or discontinuations were reported.
CONCLUSION: CT1812 improved established EEG markers of spontaneous brain activity (spectral power, functional connectivity) in patients with mild-to-moderate AD, suggesting improved neuronal/synaptic function within a 4-week timespan.
Key words: Aβ oligomer, Alzheimer’s disease, CT1812, quantitative electroencephalogram, sigma-2 receptor modulator.
Introduction
Synaptic and neuronal dysfunction and loss caused by age-dependent accumulation of synaptotoxic amyloid-beta 42 (Aβ42) oligomers may underlie cognitive decline that is typical in patients with Alzheimer’s disease (AD) (1). Accumulation of Aβ42 leads to formation of oligomers, which bind to receptor sites on the surface of neuronal synapses (2, 3). After binding, these oligomers alter membrane trafficking rate and reduce surface expression of neuronal receptors that are critical for maintaining synaptic plasticity (4, 5). This leads to failure of long-term potentiation, spine loss in neurons, and impaired cognitive performance that progresses throughout the course of AD (6-8). The sigma-2 receptor (S2R), which is encoded by transmembrane protein 97 (TMEM97), is found in different cell types including neurons. S2R regulates cell function through the progesterone receptor membrane component 1 (PGRMC1) receptor and mediates binding of Aβ oligomers to neurons in AD (2, 3).
CT1812 is a novel, first-in-class, highly brain penetrant sigma-2 receptor ligand, currently in clinical development for dementia (Alzheimer’s disease and dementia with Lewy bodies). CT1812 has demonstrated disease-modifying properties in animal models and is believed to function by preventing binding and displacing Aβ42 oligomers from receptors on brain cells (8-11). In cultured rat neurons, CT1812 antagonizes binding of Aβ42 oligomers, prevents Aβ oligomer-induced membrane trafficking changes and restores synapse protein expression (8). Chronic treatment with CT1812 restores aged AD model transgenic mouse cognitive performance to normal using multiple tests (8). Likewise, CT1812 may restore neuronal plasticity compromised by Aβ oligomers and may improve cognitive function in patients throughout the course of AD.
Various techniques can be used to study brain activity and functional connectivity, but quantitative electroencephalography (qEEG) is widely available in the hospital setting and is non-invasive, cheap, and reliable. qEEG is a neurophysiological modality that measures electrical brain activity with a high temporal resolution, providing a relatively direct measure of synaptic function. Neuronal dysfunction caused by cerebral pathology including the deposition of toxic Aβ oligomers in AD, can be detected by qEEG (12). Measures expressing local brain activity are used in the diagnostic evaluation of patients with AD in clinical care where the qEEG shows robust changes reflecting abnormalities of brain activity that may reflect disease stage (13-18).
In the prodromal phase of AD, qEEG can be normal. With progression of the disease, there is a gradual, diffuse slowing of oscillatory activity, reflected by changes of spectral power in conventional frequency bands. First, slow (theta; 4 – 8 Hz) activity increases and fast (beta, 13 – 30 Hz) activity decreases, followed by slowing and diminished reactivity of mid-range (alpha, 8 – 13 Hz) activity. Finally, very slow (delta, 0.5 – 4 Hz) power increases (15). In amyloid-positive patients who have no objective cognitive disturbances, global theta power is a predictive marker of future cognitive decline (15). Quantitative EEG measures previously have been used successfully as outcome measure in AD trials, both in the older cholinesterase inhibitor literature (19-21) and in more recent studies with different therapeutic targets, including amyloid-beta (17, 18, 22). Since global relative theta power is the most powerful early-phase diagnostic qEEG marker in AD patients (23), it was included in this study as the primary efficacy outcome measure. Several other measures including relative power in the alpha and beta bands, the global theta/alpha power ratio, posterior peak frequency and amplitude-based functional connectivity in the alpha band were also assessed (15, 24). Functional connectivity findings including reduced AECc alpha connectivity in AD were evaluated (25).
The hypothesis was tested that treatment with CT1812 can normalize qEEG patterns by improving functional connectivity and decreasing theta power relative to placebo, and that this may be an indication that CT1812 protects synapses from the toxicity of Aβ oligomers (26, 27). Normalization of the EEG means a decrease (or stabilization) of theta and increase in alpha power and functional connectivity (28). Safety was assessed and proof of mechanism of CT1812 was demonstrated in early clinical studies in healthy volunteers and patients with AD (8, 9, 29-31).
The objectives of this study were to evaluate the effect of CT1812 in restoring synaptic function in participants with mild to moderate AD using qEEG measurements and to confirm the safety and tolerability of CT1812 over a 29 day treatment course.
Methods
The study was conducted in accordance with the International Conference on Harmonization Tripartite Guideline for Good Clinical Practice, and the Guidelines of the Declaration of Helsinki. The study protocol and informed consent form were reviewed and approved by an institutional review board. All participants and their caregivers provided written informed consent prior to any study procedures.
Study Design
This was a single-center (VU University Medical Center and Brain Research Center, Amsterdam, The Netherlands), randomized, double-blind, placebo-controlled, 29-day, 2-period, crossover, Phase 2 study in adults with mild to moderate AD (EudraCT: 2019-003552-36; clinicaltrials.gov: NCT04735536). Screening procedures occurred on Days -42 to -1. Eligible participants returned to the study site at the baseline visit and were randomly assigned in Treatment Period 1 to either CT1812 or placebo daily for 4 weeks, followed by a 2-week washout period. In Treatment Period 2, patients received the alternate treatment with placebo or CT1812, and returned to the study site 12 days after the final dose of study treatment for follow-up safety and laboratory assessments. The total duration of participation in the study, including the screening period, was up to 126 days.
Patient Selection
Women and men, aged 50 to 85 years, inclusive, were eligible for inclusion if they had a diagnosis of mild to moderate probable Alzheimer’s disease dementia according to Alzheimer’s Association-National Institute on Aging (AA-NIA) 2018 criteria (32) and at least a 6-month documented history of decline in cognitive function. Participants had to use appropriate contraceptive measures during and after the trial.
To confirm the diagnosis of AD, cerebrospinal fluid (CSF) sample had to meet Aβ42 and p-tau-181 criteria: CSF Aβ42 <1000 pg/mL AND CSF p-tau 181 >19 pg/mL; OR CSF Aβ1-42 <1000 pg/mL AND p-tau-181 / Aβ42 ratio >0.020; OR CSF p-tau 181 >19 pg/mL AND p-tau-181 / Aβ1-42 ratio >0.020, as measured via an Elecsys assay. Historical CSF results were considered provided the results were consistent with the CSF thresholds required for inclusion and following discussion with the medical monitor; however, a lumbar puncture was still required as part of screening procedures.
Magnetic resonance neuroimaging (MRI) findings consistent with the clinical diagnosis of AD and without findings of significant exclusionary abnormalities were required. A historical MRI up to 1 year prior to screening was allowed as long as no clinical neurologic events occurred during this interval, which suggested a change in the MRI scan. In addition, a score of 18 to 26 on the mini-mental status examination (MMSE) (33), and no active depression and a Geriatric Depression Scale (GDS) (34) score ≤6 were required.
Participants were required to have a caregiver/study partner who had contact with the study participant for a sufficient number of hours per week to provide informative responses on study assessments, oversee administration of the study drug, and who was willing and able to participate in all study site visits and study assessments. Participants had to be living at home or in the community (assisted living acceptable) with no known history of difficulty swallowing capsules; on stable pharmacological treatment with acetylcholinesterase inhibitors or memantine for any other chronic conditions for at least 30 days prior to screening; willing to undergo apolipoprotein E (APOE) genotyping; and be generally healthy with mobility (ambulatory or ambulatory-aided, i.e., walker or cane), vision, and hearing (hearing aid permissible) sufficient for compliance with testing procedures. Participants were excluded for anything that might interfere with the conduct of the study. Detailed Inclusion/Exclusion criteria and prohibited medications are listed in Supplementary Materials.
Study Assessments
At screening, the MMSE and GDS were administered, MRI imaging and a lumbar puncture was performed, and apolipoprotein (ApoE) status was determined. Safety assessments included physical examination, vital signs (blood pressure, heart rate, body temperature), clinical laboratory testing (chemistry, hematology, urinalysis), 12-lead electrocardiogram (ECG), and adverse events. Serum concentrations of CT1812 were assessed at screening, 1 hour predose on Days 1, 8, 15, 22, 29, 44, 51, 58, 65 and 72 and 2 hours postdose on Days 1, 29, 44, and 72. CSF concentrations of CT1812 were measured at screening and 24 hours after the dose on Days 29 and 72. To assess target engagement and disease modification, plasma and CSF concentrations of Aβ42 and Aβ40 monomers, phospho-tau-217, phospho-tau-181, neurofilament light (NFL), glial fibrillary acidic protein (ng/L) were measured, and the Aβ42/40 ratio calculated. In addition, CSF concentrations of chitinase-3-like protein 1 (YLK-40, mg/L), neurogranin, neuronal pentraxin 2 (ng/L), soluble triglyceride receptor expressed on myeloid cells 2 (TREM2, ng/L), synaptosome associated protein 25 (SNAP25, ng/L); total tau (mg/dL), and vesicle-associated membrane protein 2 (VAMP2, ng/L) were measured.
Quantitative EEGs were recorded on OSG digital equipment (BrainRTTM; OSG BV, Rumst, Belgium) with Ag-Cl electrodes. An EEG assessment was conducted during a 15-minute task-free session with closed eyes on Day 1 and on Day 29 of Treatment Period 1, at Days 44 and 72 (Day 1 and Day 29 of Treatment Period 2), and at the 84 day follow-up. Twenty-one channels of the standard 10-20 system were recorded: Fp2 / Fp1, F8 / F7, F4/ F3, A2 / A1, T4 / T3, C4 / C3, T6 / T5, P4 / P3, O2 / O1, Fz, Cz, Pz, an ECG– and a respiration channel. Raw data were recorded using a montage with Fz as the common reference electrode. Electrode impedance was checked before and after the recording and was kept below 5 Kohm. Filter settings were high pass 0.16 Hz, low pass 70 Hz, and no notch filter. A sample frequency of 512 Hz and analog-to-digital conversion precision of 12 bit was used. Care was taken by an experienced EEG technician, who was blinded to treatment condition during the recording, to keep participants in an eyes-closed but vigilant condition, and to minimize any other artifacts. Five 8.2 s epochs of eyes-closed artifact-free data (containing no eye blinks, muscle artifact, slow eye movements, or EKG-artifacts) were selected from each EEG recording based on visual inspection of the data by a trained EEG technician.
The primary qEEG endpoint was global relative theta power (4-8 Hz), the fraction of total oscillatory activity in all cortical regions accounted for by theta wave frequency. Secondary qEEG measures included global relative alpha (8-13 Hz) and global relative beta (13-30 Hz) power, theta/alpha power ratio, posterior peak frequency (i.e., the dominant frequency of the power spectrum between 4 – 13 Hz in the parieto-occipital region), and global functional connectivity assessed with Amplitude Envelope Correlation (AEC-c) in the alpha band (27, 35-37). Details provided in the Supplementary Material.
Cognition and global functioning were assessed at baseline and Days 29, 44, and 72 with the Alzheimer’s disease assessment scale–cognition subscale (ADAS-Cog-14) (38) and Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change (ADCS-CGIC) (39). The Columbia Suicide Severity Rating Scale (C-SSRS) (40) was administered at screening and baseline and Days 29, 44, and 72. Plasma drug concentrations were measured predose on Days 1, 8, 15, 22, and 29 and postdose on Days 1 and 29. CSF concentrations were measured predose on Day 29.
Statistical Analysis
For the primary qEEG endpoint, global relative theta power, the assumed within-subject standard deviation (SD) was equal to 3%. Based on the use of a two-sided one-sample (within-subject) comparison between CT1812 and placebo at the alpha=0.05 level of significance, a sample size of 16 participants provided 90% power to detect a mean difference between treatments of 2.5%. Standard non-parametric statistical analyses were used to examine the relationships between CT1812 dose and PK parameters. Pharmacodynamics markers in CSF and CT1812 concentrations in serum and CSF were summarized using descriptive statistics.
Results
Of 34 patients who were screened, 16 were randomized to study treatment, and 15 completed the study. One patient discontinued from the crossover CT1812/placebo group for withdrawal of consent (death in the family). At baseline, mean (SD) age was 66.4 (7.9) years, 50% were female, and mean (SD) time since diagnosis of AD was 1.14 (1.0) years (Table 1). At baseline, mean (SD) MMSE was 21.1 (2.4), ADAS-Cog14 was 30.2 (6.9), and Amsterdam IADL was 52.6 (5.1). All participants had an AA-NIA diagnosis of mild AD, and 6 of 16 patients and 5 of 16 patients were hetero- or homozygous positive for ApoE ε4.
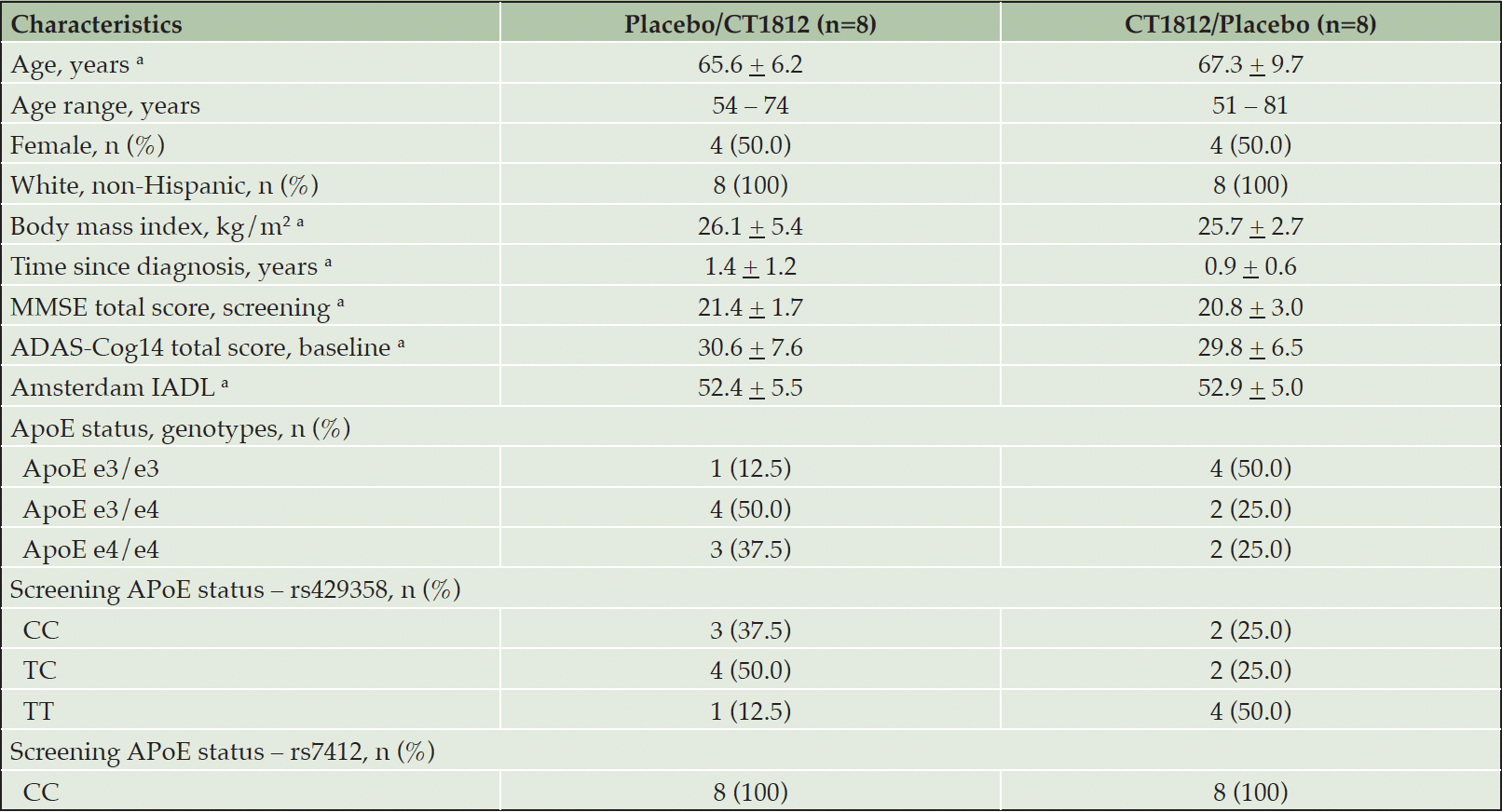
Table 1. Baseline characteristics (safety population)
a. Mean + standard deviation
Quantitative Electroencephalogram
Following treatment with CT1812 for 4 weeks, a non-significant reduction (LS mean [standard error] difference: -0.021 [0.013]; p=0.123) was observed in global relative theta power (Figure 1, Table 2). A consistent trend for decreases in relative theta power also were observed in frontal, temporal, posterior (parietal and occipital), and central brain regions (Figure 2, Table 2). The change observed in the central region was statistically significant (LS mean [SE] difference: -0.035 [0.011]; p<0.006). A nominally significant (LS mean [SE] difference: 0.016 [0.007]; p=0.034) improvement in global alpha AEC-c was observed with CT1812 (Figure 1, Table 2). A numerically favorable treatment difference (LS mean [SE] difference: 0.026 [0.017]; p=0.149) was observed for increases in global relative alpha power with CT1812 (Figure 1, Table 2).
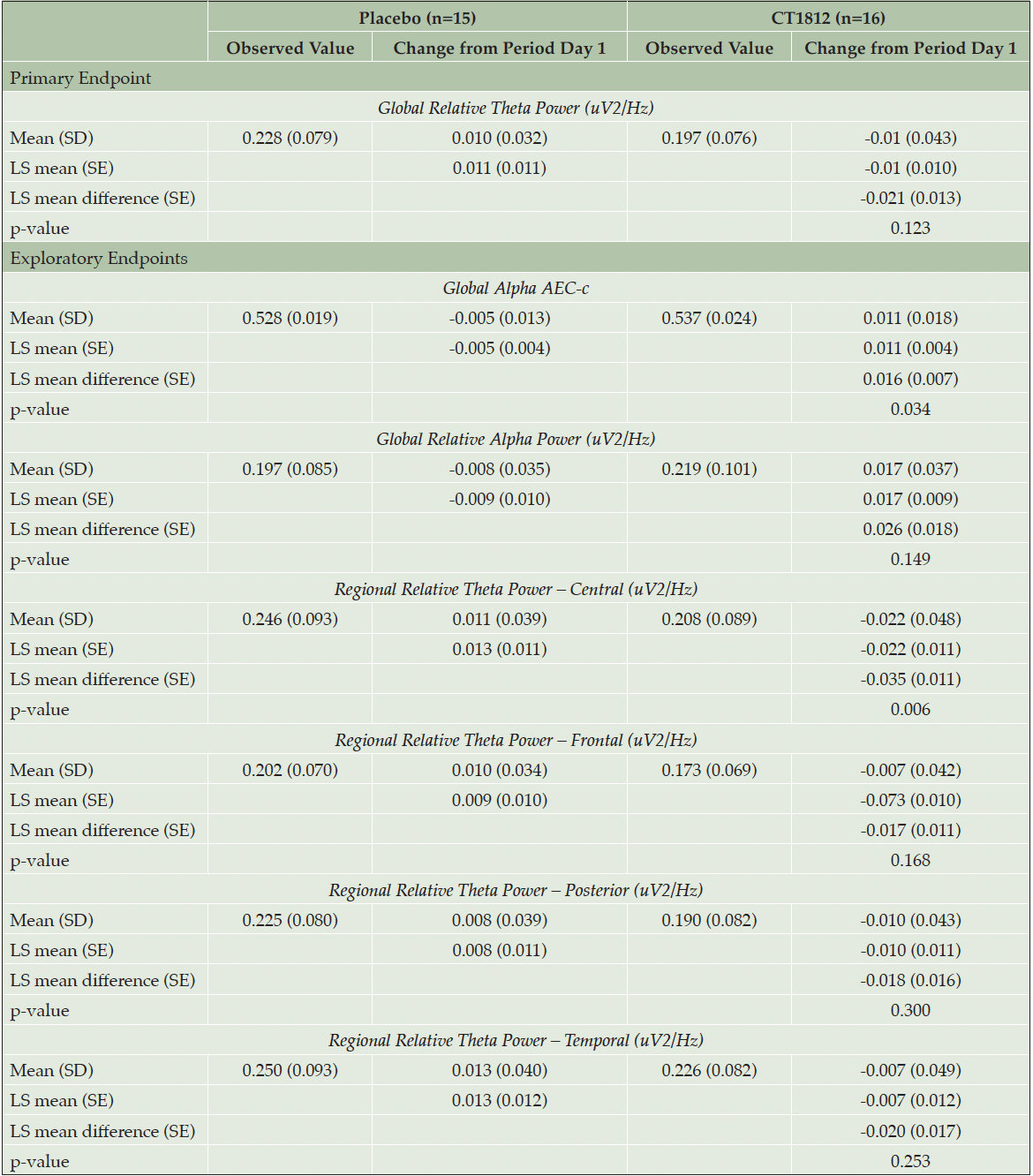
Table 2. Quantitative EEG results for primary and exploratory endpoints (safety population)
Least square (LS) mean, standard errors (SE), and p-value are estimated using a linear mixed model with fixed effects for treatment group (CT1812, placebo), sequence, and period, and a random effect for subject within sequence.
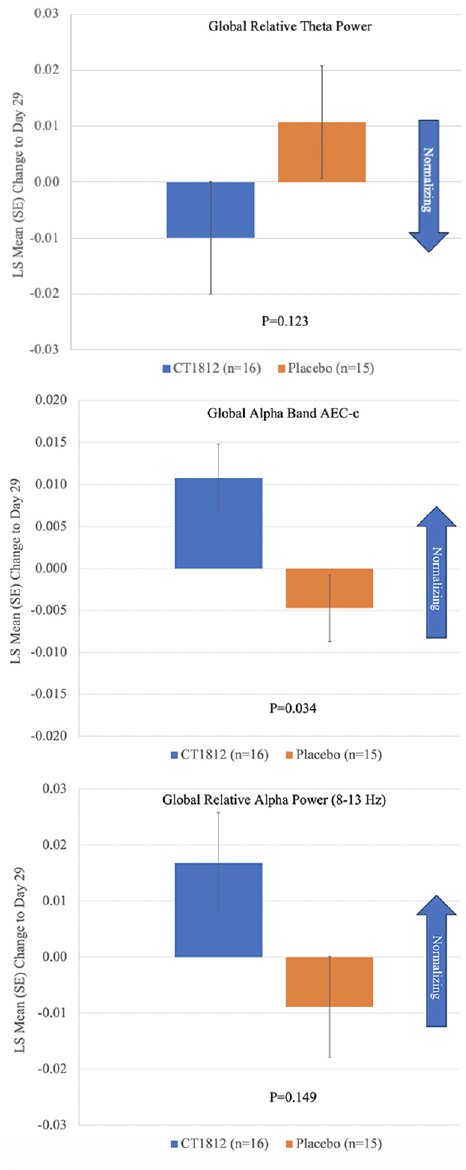
Figure 1. Mean change from baseline for Global Relative Theta Power, Global Alpha AEC-c, and Global Relative Alpha Power
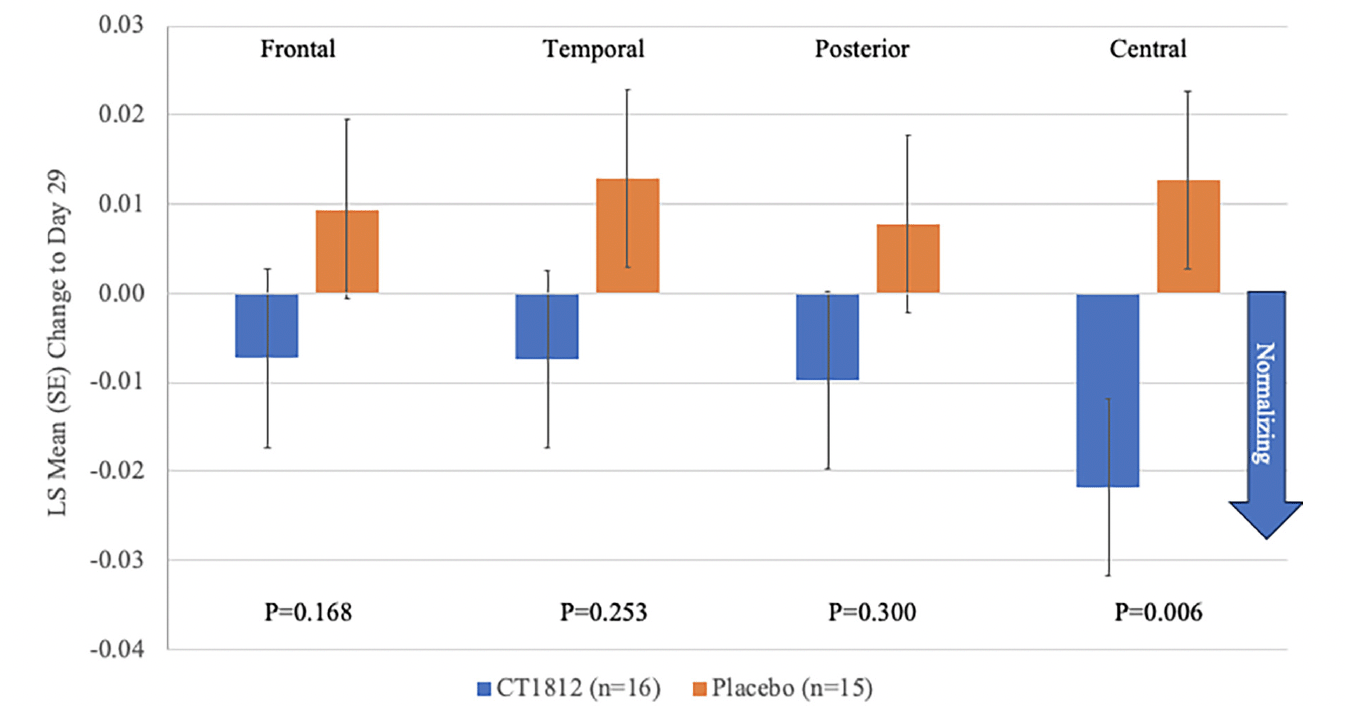
Figure 2. Mean Change from Baseline for Regional Relative Theta Power
Plasma and CSF concentrations of exploratory pre-specified biomarkers were relatively unchanged from baseline to Day 29 (Supplemental Tables 1 and 2). Mean (SD) ADAS-Cog-14 scores were 30.1 (7.2) and 29.9 (7.6) at baseline with placebo and CT1812 and mean (SD) change to Day 29 was -0.8 (5.2) with placebo and -0.7 (6.3) with CT1812 (LS mean difference 0.2, 95% CI: -2.7, 3.0, p=0.894). No significant differences were observed between CT1812 and placebo for cognitive or functional outcomes (Supplemental Table 3). At Day 29, mean (SD) ADCS-CGI-I scores were 4.0 (0.76) with placebo and 4.0 (0.73) with CT1812; all 16 patients with CT1812 had minimal or no change as did 14 of 15 patients with placebo. No differences between groups were apparent for Amsterdam IADL and the NTI Battery. Mean (SD) predose plasma concentrations of CT1812 were 25.5 (25.0), 14.9 (7.8), 13.8 (8.0), and 14.1 (9.0) ng/mL on Days 8, 15, 22, and 29, respectively. On Day 29, mean (SD) predose CSF concentration of CT1812 was 0.41 (0.25) ng/mL.
Safety/Tolerability
Mild or moderate treatment-emergent adverse events (TEAEs) occurred in 11 patients with CT1812 and 6 with placebo (Table 3). The most common AEs were nausea, diarrhea, hematoma, and procedural headache. No severe or serious AEs were reported, and no deaths or discontinuations due to TEAEs were reported. Six TEAEs (diarrhea, nausea, and vomiting with placebo; diarrhea, nausea, and hepatic enzyme increased with CT1812) were judged to be treatment-related. One patient experienced an increase in alanine aminotransferase levels that was twice the upper limit of normal and reported as a TEAE. The TEAE was of mild severity and possibly related to CT1812, but treatment was continued.
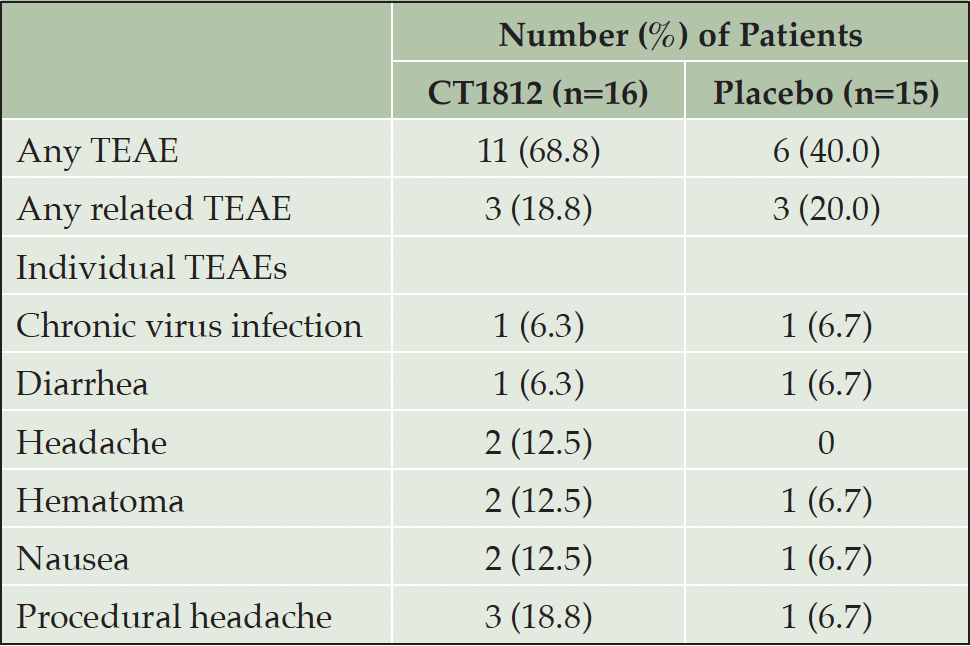
Table 3. Incidence of treatment-emergent adverse events (TEAE) occurring in more than one patient (safety population)*
* Single TEAEs occurring either group: CT1812 – atrial fibrillation, bone contusion, first-degree burn, hepatic enzyme increased; placebo – first-degree burn, pneumonia, post-procedural contusion, vomiting
No clinically relevant changes were observed from baseline to Day 29 for hematology testing or urinalysis. On the C-SSRS, suicide ideation or suicide behavior were reported in a single patient during treatment.
Discussion
In this study, qEEG was used to determine the effect of CT1812 on AD pathophysiology in the brain. Normalization would indicate a treatment effect, as this does not happen spontaneously during the disease, and we found a positive effect on oscillatory brain activity after only 4 weeks of CT1812 treatment. Results from this study support improvement in the neurophysiology of AD with CT1812. Positive, consistent trends were observed with CT1812 for the first three ranked outcomes measures (global relative theta power, global alpha AEC-c, and global relative alpha power). A consistent reduction in global and regional theta power indicates a positive trend toward normalizing brain activity with CT1812. Global alpha AEC-c reflects the ability of the brain to communicate and exchange information between brain regions and this ability is reduced in those with AD (25, 27). In this study, global alpha AEC-c was significantly improved with CT1812. In this 29-day study, CT1812 was well tolerated with only mild to moderate AEs, no serious or severe AEs, and no discontinuations or deaths attributable to CT1812.
Our results are consistent with previous trials that used resting-state qEEG outcome measures (17,25). In this 4-week study, we observed the pre-specified hypothesized changes. We observed a non-significant but consistent trend in the direction of changes that was similar to a previous study with qEEG in patients with mild or moderate AD treated for 5 days with rivastigmine (21). Of note, EEG demonstrated trends for improvement but no differences were detected in CSF and plasma biomarkers over this short treatment course. While these findings might result from a mechanistically distinct effect of CT1812, the favorable effect could be more general to a class of synaptoprotective drugs. Thus, EEG can be considered an important downstream surrogate marker of improvement.
Limitations of this study include a small sample size and a short duration of follow up. It should be noted that this study was intended to further explore the mechanism of action of CT1812. In addition, the design of the study was not adequate for showing improvements in functional and cognitive outcomes and biomarkers of AD. We focused on resting-state data, which is important given previous results, but task data may provide complementary insights. Further, qEEG mainly reflects cortical activity, while in AD, subcortical activity (e.g., hippocampal) also is important. Magnetoencephalography (MEG) can yield more detailed changes, including from deeper regions of the brain, and a single-site study with MEG would be feasible (35).
In conclusion, results from this study showed that resting state qEEG improved after CT1812. Although qEEG is an exploratory endpoint in patients with AD, substantial evidence suggests that qEEG can detect changes in both whole-brain and regional electrical patterns that are impaired in AD. Results from this study suggest that CT1812 has an effect on neurophysiological endpoints, and contribute to the growing body of preclinical and clinical evidence of the benefits of CT1812. Consistent trends for improvement were observed for pre-specified qEEG measures with significant treatment differences noted for global alpha AECc and central relative theta power. Thus, the results from this study provided encouraging preliminary results of an impact of CT1812 on brain activity in patients with mild-to-moderate AD. Larger proof-of-concept studies with CT1812 are ongoing in patients with early AD (START, NCT05531656), mild-to-moderate AD (SHINE, NCT03507790) (41), and dementia with Lewy bodies (SHIMMER, NCT05225415) that will evaluate the potential role of CT1812 as a disease-modifying therapy in patients with AD and other neurodegenerative disorders.
Acknowledgements:The authors would like to acknowledge the editorial assistance of Richard Perry, PharmD in the preparation of this manuscript, under the direction of the authors, which was supported by Cognition Therapeutics, Inc., Pittsburgh, PA.
Funding: Funding for this study was provided by the National Institute of Aging (RF1AG058710; U.S. NIH Grant/Contract) and was conducted by Cognition Therapeutics, Inc., Pittsburgh, PA. The sponsor together with authors were involved in the design and conduct of the study as well as the collection, analysis, and interpretation of data, in the preparation of the manuscript. All authors reviewed and approved the manuscript for submission.
Disclosures: SC is a former employee of Cognition therapeutics and received grant support from the National Institute on Aging (grant number NIA AG058710). PS reports receiving personal fees from Novo Nordisk outside the submitted work and is a full time employee of EQT Life Sciences. WdH reports receiving support as an EEG vendor from Cognition Therapeutics, Vivoryon, Cervomed, Treeway, and Immunobrain. AOC is an employee of and shareholder. AOC reports grants, personal fees, non-financial support and other from Cognition Therapeutics during the conduct of the study. AOC has patent number PCT/US2024/020265 pending. MEH is an employee of and shareholder in Cognition Therapeutics, Inc. MEH reports grants from NIH, and has a patent PCT/US2024/020265 pending. MG is an employee of Global R&D Partners, LLC, receives fees from Cognition Therapeutics, and owns stock in Cognition Therapeutics. EV and ES has nothing to disclose.
Author contributions: Study hypothesis and design: WdH, SC, PS. Analysis and interpretation: EV, WdH, ES, MH, MG, AOC. Supervised study: EV, AOC. Drafted manuscript: WdH, AOC. Reviewed and revised manuscript for intellectual content: EV, WdH, ES, MH, SC, PS, MG, AOC
Data sharing statement: All data for this study are reported in the manuscript and Data Supplement. Additional data may be available from the sponsor upon the written request.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Li S, Selkoe DJ. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J Neurochem. 2020;154(6):583-597. doi: 10.1111/jnc.15007. PMID: 32180217
2. Izzo NJ, Staniszewski A, To L, et al. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS One. 2014;9(11):e111898. doi: 10.1371/journal.pone.0111898.
3. Izzo NJ, Staniszewski A, To L,. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity PLoS One. 2014;9(11):e111899. doi: 10.1371/journal.pone.0111899.
4. Hsieh H, Boehm J, Sato C, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52(5):831-43. doi: 10.1016/j.neuron.2006.10.035.
5. Lacor PN, Buniel MC, Furlow PW, et al. A oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27(4):796 – 807. doi: 10.1523/JNEUROSCI.3501-06.2007.
6. Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the alzheimer amyloid-protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27(11):2866 – 2875. doi: 10.1523/JNEUROSCI.4970-06.2007.
7. Zempel H, Luedtke J, Kumar Y, et al. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013;32(22):2920-37. doi: 10.1038/emboj.2013.207.
8. Izzo NJ, Yuede CM, LaBarbera KM, et al. Preclinical and clinical biomarker studies of CT1812: a novel approach to Alzheimer’s disease modification. Alzheimers Dement. 2021;17(8):1365–82. doi: 10.1002/alz.12302.
9. LaBarbera KM, Sheline YI, Izzo NJ, et al. A phase 1b randomized clinical trial of CT1812 to measure Aβ oligomer displacement in Alzheimer’s disease using an indwelling CSF catheter. Transl Neurodegener. 2023;12(1):24. doi: 10.1186/s40035-023-00358-w.
10. Lizama BN, Kahle J, Catalano SM, Caggiano AO, Grundman M, Hamby ME. Sigma-2 receptors-from basic biology to therapeutic target: A focus on age-related degenerative diseases. Int J Mol Sci. 2023;24(7):6251. doi: 10.3390/ijms24076251.
11. Rishton GM, Look GC, Ni ZJ, et al. Discovery of investigational drug CT1812, an antagonist of the sigma-2 receptor complex for Alzheimer’s disease. ACS Med Chem Lett. 2021;12(9):1389-1395. doi: 10.1021/acsmedchemlett.1c00048.
12. Bae H, Kang MJ, Ha SW, et al. Association of plasma amyloid-β oligomerization with theta/beta ratio in older adults. Front Aging Neurosci. 2023;15:1291881. Doi: 10.3389/fnagi.2023.1291881. PMID: 38106526
13. Briels CT, Schoonhoven DN, Stam CJ, de Waal H, Scheltens P, Gouw AA. Reproducibility of EEG functional connectivity in Alzheimer’s disease. Alzheimers Res Ther. 2020;12(1):68.
14. de Waal H, Stam CJ, de Haan W, van Straaten ECW, Scheltens P, van der Flier WM, Young Alzheimer patients show distinct regional changes of oscillatory brain dynamics. Neurobiol Aging. 2012;33(5):1008.e25–31. doi: 10.1016/j.neurobiolaging.2011.10.013.
15. Gouw AA, Alsema AM, Tijms BM, et al. EEG spectral analysis as a putative early prognostic biomarker in nondemented, amyloid positive subjects. Neurobiol Aging. 2017;57:133-142. doi: 10.1016/j.neurobiolaging.2017.05.017.
16. Jeong J. EEG dynamics in patients with Alzheimer’s disease. Clin Neurophysiol. 2004;115(7):1490-505. doi: 10.1016/j.clinph.2004.01.001.
17. Scheltens P, Hallikainen M, Grimmer T, et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimers Res Ther. 2018;10(1):107. doi: 10.1186/s13195-018-0431-6. PMID: 30309389
18. Scheltens P, Twisk JW, Blesa R, et al. Efficacy of Souvenaid in mild Alzheimer’s disease: results from a randomized, controlled trial. J Alzheimers Dis. 2012;31(1):225-36. doi: 10.3233/JAD-2012-121189. PMID: 22766770
19. Arjmandi-Rad S, Vestergaard Nieland JD, Goozee KG, Vaseghi S. The effects of different acetylcholinesterase inhibitors on EEG patterns in patients with Alzheimer’s disease: A systematic review. Neurol Sci. 2024;45(2):417-30. doi: 10.1007/s10072-023-07114-y. PMID: 37843690
20. Brassen S, Adler G. Short-term effects of acetylcholinesterase inhibitor treatment on EEG and memory performance in Alzheimer patients: an open, controlled trial. Pharmacopsychiatry. 2003;36(6):304-8. doi: 10.1055/s-2003-45118. PMID: 14663655
21. Adler G, Brassen S. Short-term rivastigmine treatment reduces EEG slow-wave power in Alzheimer patients. Neuropsychobiology. 2001;43(4):273-6. Doi: 10.1159/000054902. PMID: 11340368
22. van Straaten EC, de Waal H, Lansbergen MM, et al. Magnetoencephalography for the detection of intervention effects of a specific nutrient combination in patients with mild Alzheimer’s Disease: Results from an exploratory double-blind, randomized, controlled study. Front Neurol. 2016;7:161. doi: 10.3389/fneur.2016.00161. PMID: 27799918
23. Cecchetti G, Agosta F, Basaia S, et al. Resting-state electroencephalographic biomarkers of Alzheimer’s disease. Neuroimage Clin. 2021;31:102711. doi: 10.1016/j.nicl.2021.102711. PMID: 34098525
24. Babiloni C, Arakaki X, Azami H, et al. Measures of resting state EEG rhythms for clinical trials in Alzheimer’s disease: Recommendations of an expert panel. Alzheimers Dement. 2021;17(9):1528-1553. Doi: 10.1002/alz.12311. PMID: 33860614
25. Briels CT, Stam CJ, Scheltens P, Bruins S, Lues I, Gouw AA. In pursuit of a sensitive EEG functional connectivity outcome measure for clinical trials in Alzheimer’s disease. Clin Neurophysiol. 2020;131(1):88-95. doi: 10.1016/j.clinph.2019.09.014. PMID: 31759193
26. Babiloni C, Blinowska K, Bonanni L, et al. What electrophysiology tells us about Alzheimer’s disease: a window into the synchronization and connectivity of brain neurons. Neurobiol Aging. 2020;85:58-73. Doi: 10.1016/j.neurobiolaging.2019.09.008. PMID: 31739167
27. Scheijbeler EP, de Haan W, Stam CJ, Twisk JWR, Gouw AA. Longitudinal resting-state EEG in amyloid-positive patients along the Alzheimer’s disease continuum: considerations for clinical trials. Alzheimers Res Ther. 2023;15(1):182. doi: 10.1186/s13195-023-01327-1. PMID: 37858173
28. Cummings J, Kinney J, Fillit H. (Eds.). Chapter 36, in Alzheimer’s Disease Drug Development: Research and Development Ecosystem. 2022, Cambridge University Press. doi.org/10.1017/9781108975759
29. Grundman M, Morgan R, Lickliter JD, et al. A phase 1 clinical trial of the sigma-2 receptor complex allosteric antagonist CT1812, a novel therapeutic candidate for Alzheimer’s disease. Alzheimers Dement (N Y). 2019;5:20-26. doi: 10.1016/j.trci.2018.11.001.
30. Lizama BN, North NA, Pandey K, et al. An interim exploratory biomarker analysis of a Phase 2 clinical trial to assess the impact of CT1812 in Alzheimer’s disease. bioRxiv 2024.02.16.578765; doi: https://doi.org/10.1101/2024.02.16.578765
31. Rasheed A, Zaheer AB, Munawwar A, et al. The allosteric antagonist of the sigma-2 receptors-elayta (CT1812) as a therapeutic candidate for mild to moderate Alzheimer’s disease: A Scoping Systematic Review. Life (Basel). 2022;13(1):1. doi: 10.3390/life13010001.
32. Jack CR Jr, Bennett DA, Blennow K, et al; Contributors. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535-562. doi: 10.1016/j.jalz.2018.02.018.
33. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(13):189 -198. doi: 10.1016/0022-3956(75)90026-6.
34. Sheikh JI, Yesavage JA. Geriatric Depression Scale (GDS): recent evidence and development of a shorter version. In: Brink TL, ed. Clinical Gerontology: A Guide to Assessment and Intervention. New York, NY: The Haworth Press; 1986:165-173.
35. Brookes MJ, Woolrich MW, Barnes GR. Measuring functional connectivity in MEG: a multivariate approach insensitive to linear source leakage. Neuroimage. 2012;63(2):910–20.
36. Bruns A, Eckhorn R, Jokeit H, Ebner A. Amplitude envelope correlation detects coupling among incoherent brain signals. NeuroReport. 2000;11(7):1509–14.
37. Hipp JF, Hawellek DJ, Corbetta M, Siegel M, Engel AK. Large-scale cortical correlation structure of spontaneous oscillatory activity. Nat Neurosci. 2012;15(6):884–90.
38. Kueper JK, Speechley M, Montero-Odasso M. The Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog): Modifications and Responsiveness in Pre-Dementia Populations. A Narrative Review. J Alzheimers Dis. 2018;63(2):423-444. doi: 10.3233/JAD-170991.
39. Schneider LS, Olin JT, Doody RS, et al. Validity and reliability of the Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord. 1997;11 Suppl 2:S22-32. doi: 10.1097/00002093-199700112-00004.
40. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266-77. doi: 10.1176/appi.ajp.2011.10111704.
41. Pandey K, Duong D, Waybright L, et al. Pharmacodynamic effects of the S2R modulator CT1812 in Alzheimer’s disease patients observed in a meta-analysis of CSF proteomes from SPARC and SHINE Part A. Poster presented at: AD/PD™ 2023 International Conference on Alzheimer’s and Parkinson’s Diseases
© The Authors 2024